FDA官方深度解读:从最新申报数据与483检查看冻干工艺趋势、控制缺陷与未来方向
本文作者团队来自FDA药品评估与研究中心监管人员:
Steve Y. Rhieu – 药品质量办公室(OPQ)下属药品制造评估办公室(OPMA)专家,负责冻干工艺及相关注射剂产品的CMC审评与合规评估
Maxwell Korang-Yeboah – 药品质量研究办公室(OPQR)研究员,专注于冻干等先进制造技术的研究与评估
David D. Anderson、John Arigo – OPMA资深审评员,拥有多年无菌制剂及冻干工艺现场检查与申报资料审评经验
Thomas O’Connor – OPQR专家,主导制药过程建模与新兴技术评估项目
Rakhi Shah – OPQ高级政策专家
该团队代表了FDA在冻干工艺科学审评、合规监管与新兴技术推进方面的核心力量。其工作直接衔接CDER内部的新兴技术计划(ETP)审评流程,并参与制定相关行业指南。
FDA通过OPQ下设的多个专家工作组(如无菌工艺工作组、先进制造技术工作组)系统性推进冻干领域监管科学与行业标准的升级。
本研究报告是这一监管科学体系的最新产出。
冷冻干燥是生物制剂、高端无菌注射剂等不稳定药物产品的关键技术之一。COVID-19大流行期间,mRNA疫苗的冻干化开发凸显了该技术在公共卫生应急中的战略价值。
伴随FDA“21世纪药品质量计划”及药品质量办公室(OPQ)的持续推动,冻干工艺正从经验驱动向科学化、数字化与连续化制造演进。
01、监管视角下的研究背景与方法论
本研究采用双重数据源交叉分析的监管科学方法:
申报资料分析:系统审阅2020-2023年间FDA接收的162份包含冻干剂型的申报资料(24项NDA、118项ANDA、20项BLA),聚焦其CMC章节中的工艺开发、控制策略及放大方案
检查数据回溯:分析2015-2019年间进行冻干生产的药品生产基地的201份FDA检查报告,特别是其中引出的Form FDA 483观察项,以识别系统性合规缺陷
数据全部来源于FDA内部审评与检查数据库,为监管机构基于真实世界审评与检查数据的趋势研判能力的体现。
这种“申报-检查”联动分析是FDA OPQ用于识别行业共性短板、精准发布合规指南的重要方法论。
02、申报资料呈现的工艺控制策略图谱
终点判定方法的选择:直接反映企业对工艺的理解深度
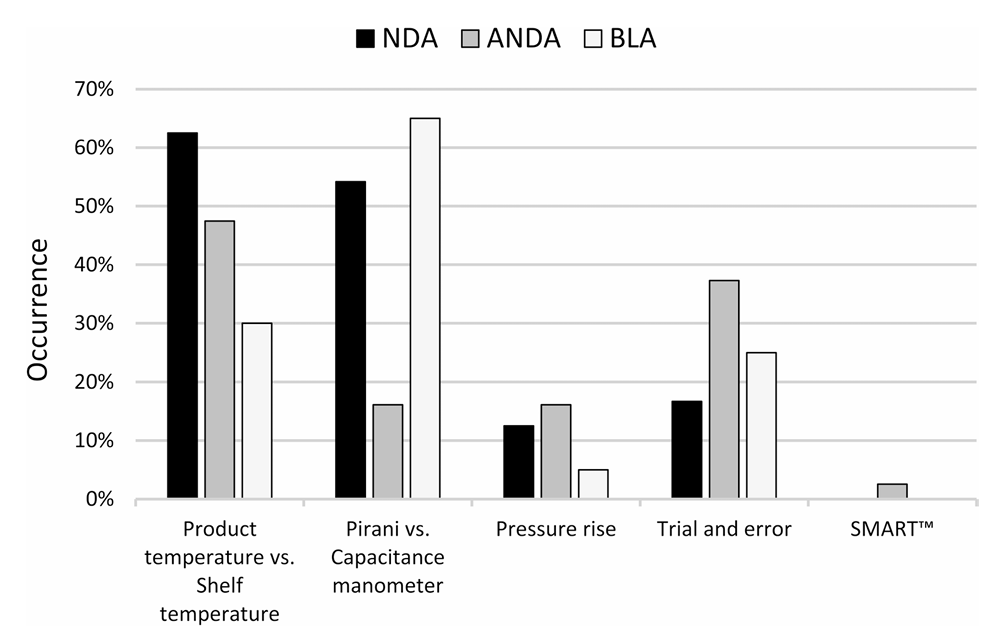
图1:从所审查的24份NDAs、118份ANDAs及20份BLAs中观察到的、用于确定干燥终点的方法
数据显示:
62.5%的NDA与47.5%的ANDA采用 “产品温度接近板层温度”作为一次干燥终点判据,该方法判断的终点常常提前,导致一次干燥时间不足;
压力上升测试(PRT)与电容/皮拉尼对比压力监测在BLA中应用率达55%,体现了生物制剂企业对PAT工具的更高接受度;
SMART™等基于MTM的先进控制策略整体应用率不足3%,且主要见于ANDA,揭示先进PAT在商业化生产中的渗透仍面临障碍
令人关注的是,“试错法”工艺开发在ANDA中占比仍高达37.3%,这反映出部分仿制药企业在基于科学和风险的工艺开发(QbD)实践上仍有提升空间。
在商业化放大策略上,多数申报遵循FDA指南采用1.5-10倍的放大系数,并倾向于在放大时保持设备型号与关键工艺参数不变。
然而,仅不足5%的NDA/ANDA申报资料中包含了将PAT工具整合进商业化生产的工艺控制方案,显示出 “开发-生产”之间技术转移的断层。
03、483检查观察项揭示的六大合规痛点
通过对201份EIR中104份涉及冻干的483观察项进行归类分析,FDA专家团队识别出当前行业在冻干生产合规中的六大高风险领域:
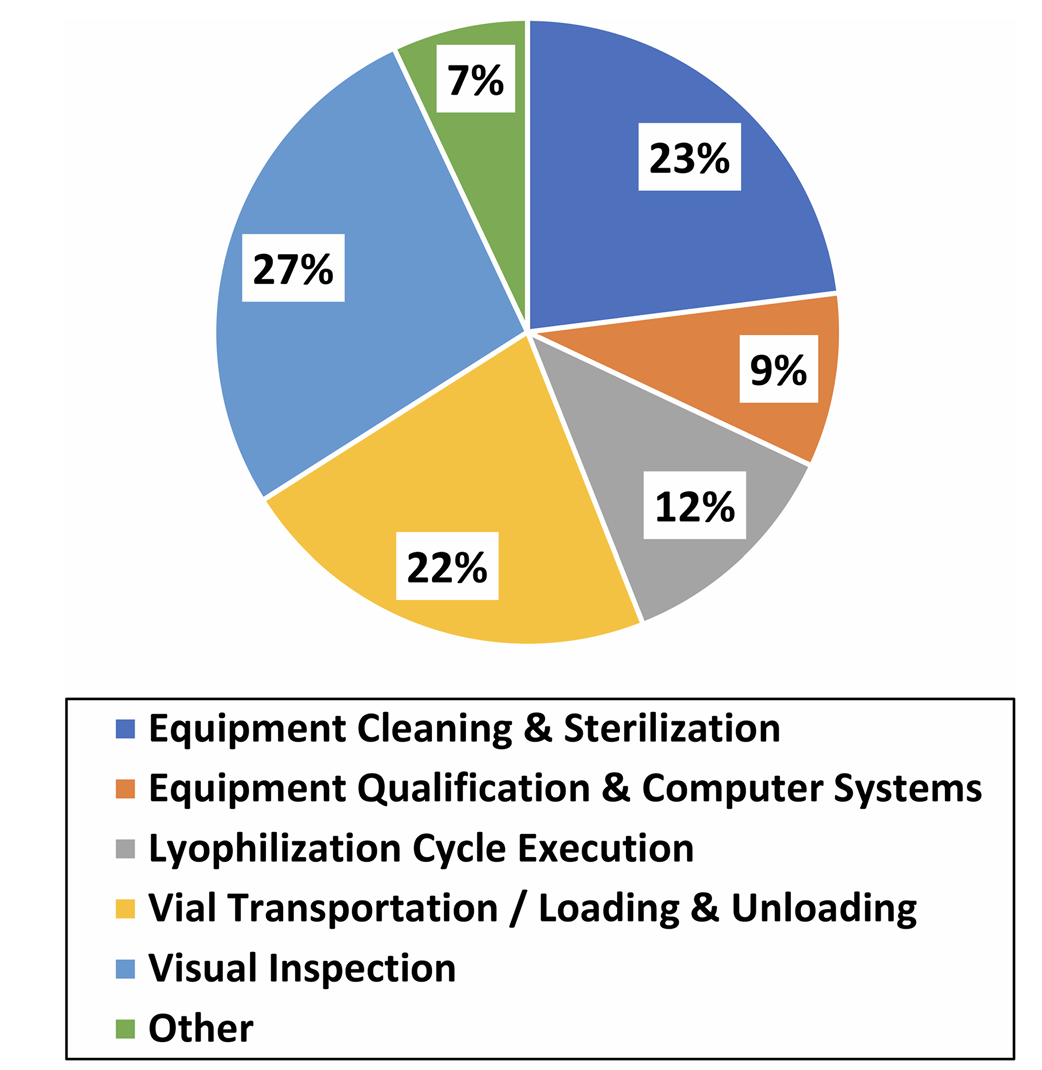
图二:2015-2019年间冻干相关483观察项分布(基于104份含缺陷的EIR)
1、视觉检查程序(占比27%)
这是最常见的缺陷领域,具体问题包括:
(1) 用于人员培训的缺陷标准品库不完善或不具代表性,导致检查标准不统一。
(2) 产品质量投诉、AQL抽样检验失败、留样及稳定性样品中发现的缺陷,均指向检查人员培训不足,未能有效识别缺陷。
(3) 未遵循USP相关建议,例如:未控制每支西林瓶的检查时间、未考虑操作员疲劳因素、光照强度不达标、未使用推荐的黑白检查背景等。
2、设备清洁、灭菌及无菌操作(占比23%)
此类别的高频问题突显了无菌保障环节的脆弱性:
(1) 对用于压塞后反填气体(如氮气)的除菌过滤器,未进行完整性测试。
(2) 人员在执行环境监测前才进行手部消毒,监测行为本身可能已受到污染。
(3) 非活性粒子探头和沉降碟的放置位置不当,无法有效监控关键区域。
3、部分加塞瓶转运与装载过程(占比22%)
从灌装线到冻干机的衔接是污染高风险点:
(1) 在从高效空气过滤器(HEPA)小车卸载托盘或向冻干机内装载托盘时,操作不当破坏了“首过空气”的保护原则。
(2) 未进行或未充分进行烟雾研究,以可视化并验证转运路径上的气流形态和洁净度保持情况。
4、冻干循环执行与监控(占比12%)
工艺控制本身的问题包括:
(1) 未基于产品特异性研究来定义关键工艺参数(CPP)的限度。
(2) 缺乏对历史冻干循环数据的定期回顾与分析。
(3) 没有产品专属的开发数据来支持所设定的冻干工艺。
(4) 对生产过程中出现的工艺偏差调查不充分、不彻底。
5、设备与计算机系统验证(占比9%)
数据可靠性相关问题日益受到关注:
(1) 用户访问权限未按角色进行严格限制,存在数据篡改风险。
(2) 存在数据检索困难或无法完整检索的问题,影响追溯性。
6、质量体系关联缺陷(占比7%)
体系层面的不足会影响对冻干工艺的持续监控:
(1) 未对产品投诉进行趋势分析。
(2) 与冻干工艺相关的调查或纠正与预防措施(CAPA)不充分。
视觉检查成为483观察项的最高频来源,这提示企业需重新审视其视觉检查体系的科学性与合规性。
FDA建议企业参考USP标准及Patel等人(2017)发表的冻干饼外观可接受标准指南,建立客观、一致的检查标准。
04、四大新兴技术:从科学概念到监管接纳
4.1 受控冰核技术:从异质性控制到工艺强化
传统冻干的随机成核机制导致批次内过冷度差异可达10°C以上,进而引起干燥速率与产品质量属性的瓶间差异。
CIN技术通过压力震荡、冰雾引入、负压法等物理手段,使全部药瓶在设定温度点附近同步成核。
FDA OPQR的研究表明,成功实施CIN可使一次干燥时间缩短20-30%,并显著提升产品饼状结构的均一性。
自2020年以来,已有1项NDA与1项BLA获得包含CIN技术的工艺批准,另有3项申请正通过ETP进行前期技术论证。
4.2 PAT工具集群:从离线检测到实时放行
FDA对于PAT在冻干中的应用持明确鼓励态度,但其商业化实施需跨越三大障碍:
技术障碍:传感器与冻干机的兼容性(如TDLAS需光学视窗)、无菌保障挑战(探头引入可能破坏无菌屏障)
监管障碍:实时监测数据的GMP合规性、PAT方法验证的复杂性
商业障碍:对于已上市产品的变更管理成本、仿制药企业的价格竞争压力
对比压力监测(双真空计法)成为目前商业化应用最广的PAT工具,其通过电容规(绝对压力)与皮拉尼规(气体组成依赖性压力)的读数差异识别一次干燥终点,无需侵入产品环境。
TDLAS与无线温度传感器(如TEMPRIS)正从研发向生产场景渗透。
TDLAS可直接测量水蒸气质量流量,为干燥动力学建模提供关键输入;而无线传感器解决了传统热电偶在自动装载系统中的实操难题,已在部分欧洲生产线实现商业化应用。
4.3 过程建模:从经验放大到数字孪生
一次干燥模型基于传质-传热第一性原理,已成为工艺开发与放大的重要辅助工具。模型的关键输入包括:
瓶底热传导系数(Kv):考虑辐射、传导、对流及边缘效应
干燥层阻力(Rp):与饼层孔隙结构、冰晶形态密切相关
设备能力边界:冷凝器捕获能力、真空系统稳定性
FDA于2023-2025年连续发布关于计算模型可信度评估的指南文件,为模型在监管申报中的应用提供框架。行业案例显示,采用模型可减少30-50%的工艺开发实验批次,并在技术转移中更好地预测规模效应。
4.4 连续与微波辅助冻干:下一代制造范式探索
连续冻干:通过旋转冻干或悬浮瓶连续处理系统,实现单位剂量产品的“一个流”生产。其核心优势在于消除批内瓶间差异,但设备设计与无菌控制面临全新挑战。
微波辅助冻干:利用微波的体积加热效应大幅缩短干燥时间。FDA CDER近期资助专项研究,旨在建立MAFD的传热-传质模型及配方介电特性数据库,为后续技术产业化奠定科学基础。
目前尚未有基于连续或微波冻干的商业化申报,但这两种技术代表了冻干制造范式向更高效、更个性化方向演进的重要探索。
05、无菌保证:冻干工艺的特殊控制维度
冻干机作为无菌生产线的延伸,其无菌控制需覆盖 “设备-工艺-环境” 全链条:
设备灭菌:SIP验证需关注温度分布均匀性、生物指示剂挑战及最差条件模拟
环境衔接:从灌装线到冻干机的转运路径必须维持ISO 5/A级环境,并应通过烟雾研究可视化气流形态
工艺模拟:培养基模拟试验应覆盖部分加塞瓶转运、冻干机装载/卸载、真空破空等所有高风险手动操作步骤
FDA检查中常见的缺陷是工艺模拟使用的冻干机型号与商业化生产不一致。监管机构强调,模拟应尽可能还原实际生产的全部条件,包括设备类型、装载方式及停留时间。
06、监管申报策略与合规升级路径
6.1 针对新兴技术申报的监管建议
早期互动:通过FDA ETP在IND/pre-IND阶段就CIN、连续冻干等创新技术与审评部门沟通
基于风险的验证:识别新技术引入的潜在失效模式(如冰雾分布不均、微波热点形成),并设计针对性控制策略
对比数据:提供与传统工艺的对比研究数据,证明新技术在质量、效率或可控性方面的优势
6.2 针对483常见缺陷的整改方向
视觉检查体系升级:采用自动化视觉检查系统减少人为变异(我本人感觉灯检机这个设备还没进化到这么智能,可以对冻干产品进行高效检查,尤其是外观不那么完美的产品),并建立基于产品特性的缺陷分类库
无菌衔接控制强化:实施全程粒子监测,并对转运操作进行定期再验证
数据驱动工艺监控:建立冻干循环数据的统计过程控制图表,实现早期异常预警
07、未来展望:监管科学与产业创新的协同进化
冻干工艺正处从“艺术”到“科学”的关键转型期。FDA通过本研究向行业传递明确信号:
1、数据驱动的监管理念:企业应系统收集并分析工艺数据,用证据证明工艺的稳健性与产品质量的一致性
2、技术中立的标准框架:监管关注点将从“如何生产”转向“产品质量是否受控”,为新技术应用开辟空间
3、全生命周期管理:从研发到商业化生产的技术转移透明度,将成为监管审评的重点考量因素
FDA OPQ将继续通过ETP、指南制定与科研项目,搭建行业与监管的技术对话平台。对于有志于冻干技术创新的企业,建议:
关注ICH Q13(连续制造) 及FDA新兴技术指南的最新进展
主动参与USP冻干相关通则的修订与标准建立
探索数字化工具(如过程模型、数字孪生)在工艺开发与合规中的应用
本文基于FDA CDER OPQ官方研究,由直接负责冻干产品审评与检查的监管专家团队撰写,代表了当前FDA在该领域的最新监管科学与合规期望。研究显示,虽然行业在冻干科学理解与控制策略上已有进步,但在视觉检查、无菌衔接、数据完整性等方面仍存在系统性合规短板。
新兴技术(CIN、PAT、连续制造)的商业化应用仍处于早期,但其在提升质量均一性、生产效率与供应链韧性方面的潜力已获监管机构认可。成功的技术采纳需要早期且透明的业界-监管沟通,以及基于风险的验证策略。
随着全球药品供应链与公共卫生需求的变化,冻干技术的创新与合规升级已成为行业必修课。FDA将通过持续的研究、指南更新与国际合作,支持这一关键制造领域的现代化进程。
作者声明:本文观点为作者个人专业见解,不代表FDA官方政策立场。
参考文献
[1] Rhieu SY, et al. AAPS Open. 2025;11:27.
[2] FDA Guidance: Sterile Drug Products Produced by Aseptic Processing (2004)
[3] USP Chapters 〈790〉, 〈1790〉
[4] Patel SM, et al. J Pharm Sci. 2017;106(7):1706-1712.
[5] ICH Q13: Continuous Manufacturing of Drug Substances and Drug Products (2023)
邵丽竹
何发
相关推荐
-

特别专题 | 连续制造如何实现从工艺开发到生产的连续化与灵活化
凭借工艺技术优势,连续化药品与营养保健品生产正日益普及。菲特公司以 FE CPS 连续处理系统为核心,作为工艺合作伙伴推动全面战略转型。
2025-12-05
-

基于 GMP 检查视角的无菌药品生产环境监测要点分析
目的:阐述无菌生产环境监测重点关注内容,分析常见检查缺陷,以期为无菌药品生产企业提升环境监测能力提供参考,同时为药品生产质量管理规范(GMP)无菌检查环境监测提供检查思路。
2025-12-04
-

底喷微丸包衣工艺参数对包衣效果的影响分析
流化床底喷微丸包衣的工作原理是借助急速上升的空气流使微丸在包衣室内悬浮流动,微丸在干燥室内上下翻动,利用喷枪借助压缩空气将包衣液雾化喷入流化床,在微丸表面形成一定厚度的膜,同时通入热空气迅速干燥上药后的微丸的包衣方法。
2025-12-04



热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多