生物制药数字化工厂 规划设计与实践
在生物制药工厂中,利用数字化和自动化控制技术,通过对生物制药设备进行集成与优化,构建生物制药生产运行的自动控制系统,提高生物生产全过程的质量控制水平,可以实现生物制药智能化。生物制药数字化,即运用现代科学技术,对生物制药各环节进行监测和控制,以达到数据安全可靠、缩短产品时间、增强生产灵活性、提高产品质量、实现节能增效的目的。
定义和质量要求
数字工厂除了包括数字化设备及生产设施以外,还应包括对生产过程的数字化管控。最重要的是要符合生物制药行业特点,满足药品生产质量管理规范(GMP)要求,满足生物制药数据的安全和完整性的要求,并根据安全条款对过程控制系统应用程序的访问进行验证,以确保安全性。为了符合法规,对各系统的访问由系统权限与安全控制要求和站点安全程序构成。数字化系统要符合制药“计算机化系统”的要求[1],FDA 21CFR Part 11 中“电子签名、电子记录”的要求[2]。
设计、建造的一般原则
坚持质量源于设计(QbD)的理念,基于风险分析,对系统全生命周期进行规划设计:
● 通盘考虑底层制药装备和制药生产过程的数字化;
● 将生产所用设备设施等数字化设备进行互联互通,为实现数字化系统与物理系统的深度集成作预留;
● 确保数字化设备与系统的合规性;
● 制定自动化控制系统的安全策略;
● 环境监测系统与楼宇控制系统是两套独立的控制系统,从系统设计到仪表安装全部采用独立的设备和仪器仪表;
● 数字化系统访问安全需要有完善的密码保护策略,防上密码控制失效。
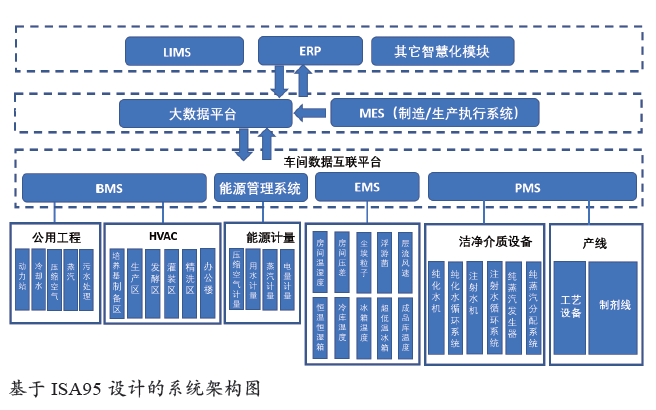
数字化单元系统架构的确定
基于QbD 理念,参照美国仪表学会(ISA)发布的S95 和S88模型[3],可以规划设计三层架构,第一层是设计建造的重点,包括生产过程监视系统(PMS)、环境监测系统(EMS)、楼宇控制系统(BMS),以及能源智能监控系统(PMS)的数字化单元,优先实现工厂的数字化。
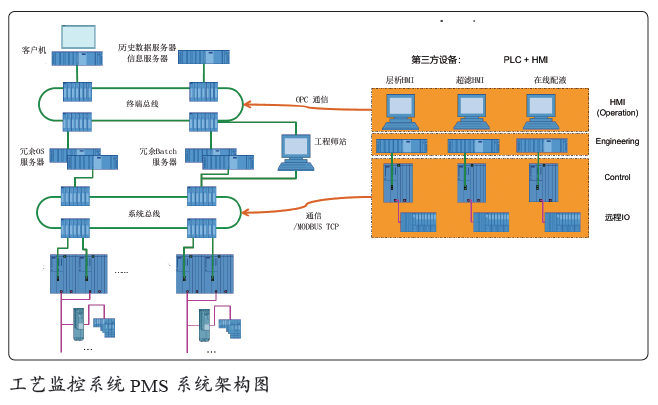
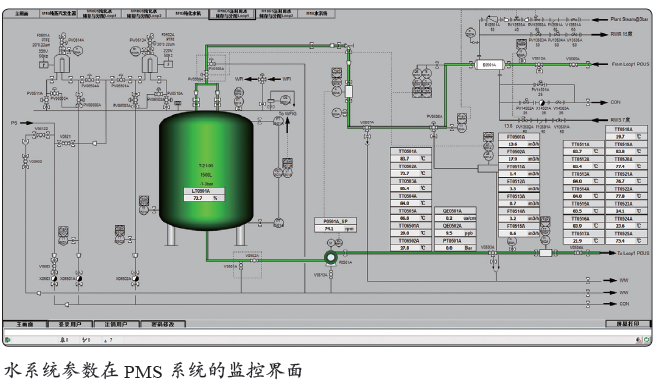
PMS系统架构及介绍
PMS 由系统服务器、冗余服务器、操作员站或人机界面(HMI)、控制器、输入/ 输出(I / O)系统、本地历史数据库存等组成。并且能够与第三方接口连接,通过以太网通信向本地的PMS 历史数据提供监控和数据采集。
PMS 工艺监控系统可以对第三方设备的数据进行采集、集成,如下所示:
● 纳滤系统集成:SIP 功能,工艺数据采集(温度、压力等)。
● 层析系统集成:Buffer 请求功能,工艺数据采集(压力、温度、流量、光度等)。
● 超滤系统集成:超滤设备CIP 循环清洗功能,工艺数据采集(进口压力、进口流量、出口流量、透过流量、温度等)。
● 洁净共用系统:工艺气体、水系统的运行参数(电导率、pH、TOC、温度、流量及压力等)。
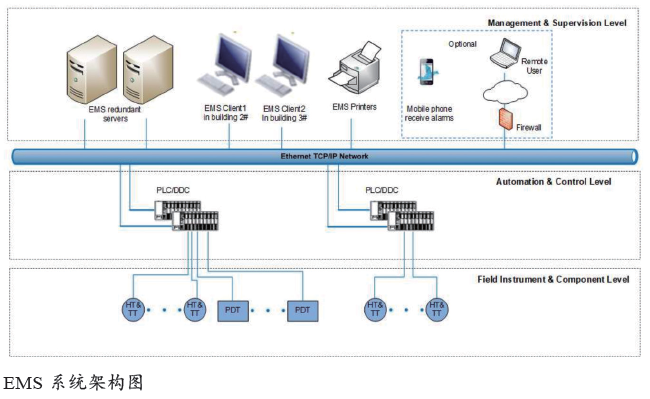
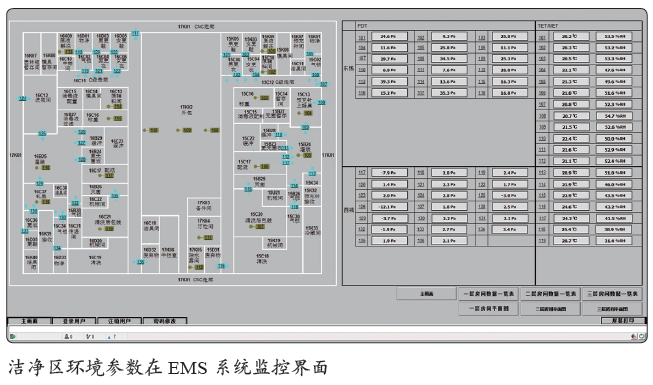
EMS系统架构及介绍
洁净厂房环境监视系统(EMS)是对GMP 环境的监测系统,是对于受BMS 控制下的生产洁净室参数的监测,当环境状态发生波动或变化时,会产生报警和记录,用于提示后期对异常状态的溯源、分析和改进。
EMS 主要对环境温度、湿度、压差、悬浮粒子和浮游菌,以及各种冰箱培养箱等的温度进行持续在线监测,并提供但不限于以下的功能:报警通知以及报警限管理;报告管理;历史数据的采集和存储审计追踪;用户管理;电子签名。
EMS 除了设计和配置上述基本功能及法规外,其功能还具有归档周期设置和维护(报警信息)打开/ 关闭两个特点。
EMS 的设备组成部分包括:监视数据、处理报警,以及与用户接口的服务器及工作站;洁净区域和关键区域的温度探头/ 传感器,如生产区域、仓库、冷库等。
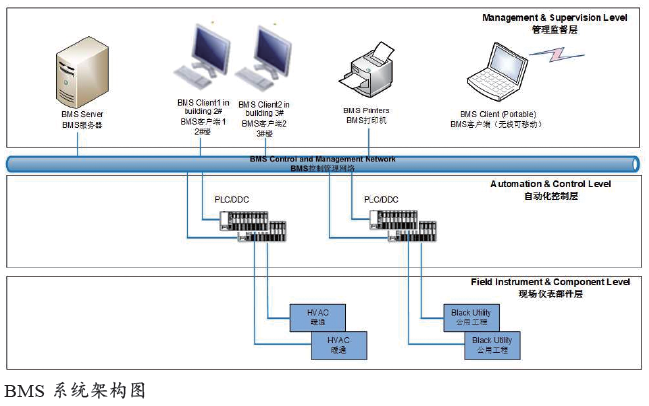
BMS系统架构及介绍
BMS 是一个基于PLC 或DDC 技术的计算机控制自动化系统,分为三层结构:
● 管理层:包括BMS服务器、客户端,以及一台网络打印机。
● PLC/DDC 控制层:独立的控制器由CPU、电源模块、通信接口和I/O 模块等组成;通信接口和I/O 模块用于采集现场仪表、现场单元或设备的信号。
● 现场仪表/ 部件层:包括仪表和执行器,这些部件安装在受控单元,并从集成的独立控制盘柜和MEPs 接收。
BMS 是对于楼宇或厂房的综合性管理系统,其功能可以完全或部分覆盖HVAC 机组控制、公用工程设备控制、房间送风排风、给排水的控制或监视,以及附属功能(如弱电系统的集成)。BMS除具备温湿度、压力控制功能外,还设计了正常工作、消毒模式、消毒排风和节能模式功能。
BMS 设计标准过程中,重点考量了以下生物制药空调系统的运行特点和要求,以实现:
● 空调系统与公用工程系统共同工作,向洁净室区域提供经空调过滤的空气。
● 空调机组与各个房间风管调节装置共同工作,为洁净室区域提供所需的压力水平。每天24 h 连续操作,可以通过BMS 手动启动/ 停止。
● 系统安全主动与本地开关设置为自动或手动。
● 将冷却盘管与加热盘管分开控制,以满足出/ 供气设定值温度,当供气管道上的供气相对湿度达到最大设定值时,对冷却盘管进行过载控制。
● 将冷却盘管与加湿器蒸汽分开控制,在设定点排出/ 供给空气相对湿度。
● 管道进房间前增加热水加热盘管,使其在设定温度下保持室温。
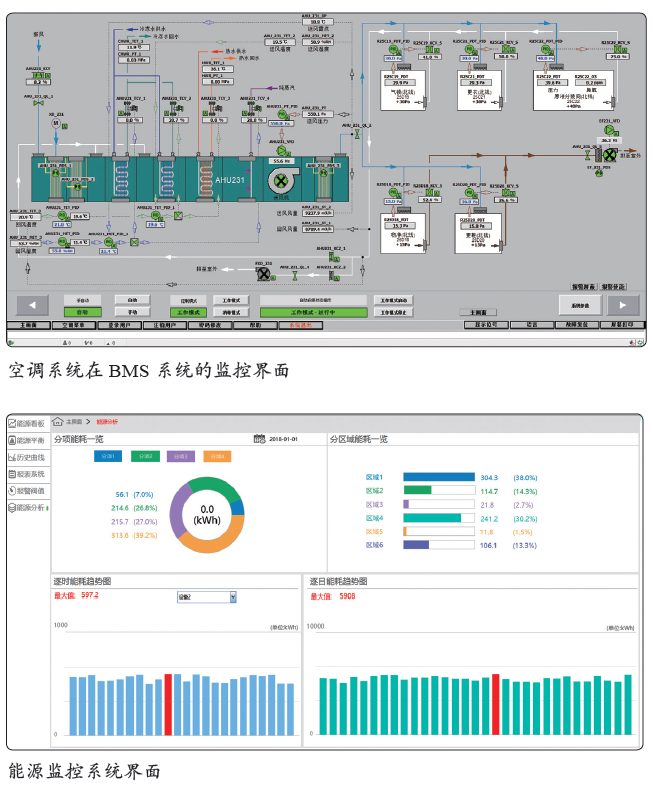
能源管理系统
生物制药工厂的水电气等能耗比较高,需要进行优化能源的管理及监控,以减少能源的浪费,达到精益生产的目标。通过对能耗及能源成本等大数据进行分析,自动进行负荷预测,调节负荷开关,达到整体能耗的最低化。能源监控系统与智能化机房相结合,并具备看板管理和多重终端查看功能。
基于灾难备份机制的高可用性数据完整性平台
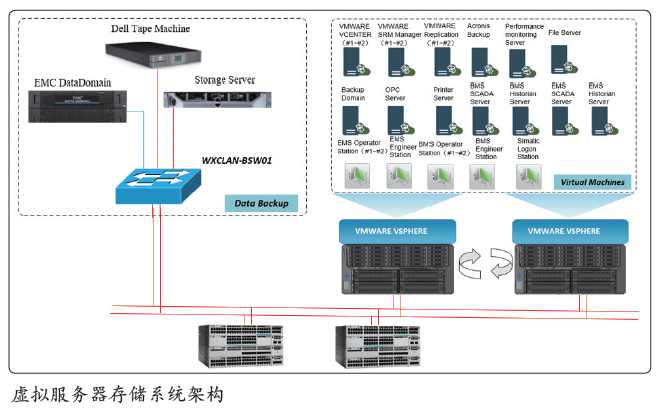
从历年FDA485.3 警告信的数据来分析,数据完整性问题引起的偏差数量最多,除了设计的系统要符合数据完整性的要求,使用虚拟化计算机系统对GMP 关键数据进行采集和保存也很重要,如配置VMWARE 虚拟化系统来实现计算资源的集群和存储。系统要求支持RAS 和BMS 等控制级应用程序,在所有区域中,部署冗余域控制以进行应用程序系统身份验证和该区域的网络设备时间同步;每天都用于数据备份,备份数据可以存储在EMC 数据上域存储单元。
各系统设计、集成特殊要求
智能化系统的第一层系统和各设备控制系统,往往是不同厂家的产品,在规划设计阶段要在URS 中提出各分、支系统的通信要求和采集、存储开放要求:
1. 供应商成套设备需要开发通信接口给第三方数据采集系统,以备采集供应商成套设备的实时数据和画面。
2. 成套设备供应商根据设备的URS 要求和FDA 21CFR Part11 的要求提供系统功能描述和自动化系统实施、验证的服务;同时能够满足控制系统接口的要求。
3. 供应商成套设备可以是一个独立的设备控制单元,或由工厂集成的工艺控制单元,并且经过FAT 测试后才能发货,并且应有打印功能,以便打印工艺相关报告。
良好的设计和集成,可以达到一些功能和效果:同步关键质量参数和生产过程参数,同步各系统信息;可以把批次配方与操作站和HMI 等无缝连接一起;把来自不同数据源的数据聚集在一张批记录报表上;提高数据的完整性,减少错误。
系统的确认和验证
智能化系统作为计算机化系统,首先应进行系统影响性评估,即用以判别系统对产品质量或生产工艺、患者安全、数据可靠性有无GMP 影响的系统层面的评估。系统的影响程度决定了进行不同程度的计算机化系统验证或者是否需要做验证。根据计算机化系统分类的不同,通过GAMP5 中GxP 影响性评估、系统软硬件分类评估、记录供应商评估结果,以及评估对于FDA 21 CFRPart 11 适用性要求,来决定是否需要进行计算机化系统验证。其次是进行风险评估,风险管理贯彻整个系统生命周期,根据基于应用软件的系统的分类(CAT3 类、4 类或者5 类)情况,确定系统的总体风险为“低风险”“中等风险”或者“高风险”。最后是使用GAMP 5 的软件类别定义方式以及相应的基本验证活动规则,来规范系统的验证活动[4]。通常对于基于软件类别3、4、5 的系统需要进行不同程度的验证,对于软件类别1 无需进行验证,但需要针对支持GMP 活动的计算机化系统的IT 基础设施进行确认。
【参考文献】
[1] 国家药品监督管理局. 中国GMP2010,附录:计算机化系统.
[2]US Food and Drug Administration (FDA) ,21 CFR Part 11 – Electronic Records, Electronic Signatures.
[3]International Society of Automation,ISA-95Enterprise-Control System Integration,2018.
[4]International Society Pharmaceutical Engineering, ISPE Good Practice Guide GAMP®5 : A Risk-Based Approach to Compliant GxP Computerized Systems, 2017.
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多