2023年获FDA批准的小分子新药及药物设计思路
小分子药物结构、机制与适应症汇总
获批的29个小分子的信息汇总如下:

1/20/2023: Brenzavvy (bexagliflozin)
1/27/2023: Jayprica (Pirtobrutinib)
基本信息:Pirtobrutinib是由Lilly公司开发(该药物最初由Redx Pharma研发,后续被Loxo Oncology以4000万美元收购,Lilly又于2019年以约80亿美元收购Loxo)的非共价BTK激酶抑制剂,用于治疗至少二线全身治疗(包括BTK抑制剂)后复发或难治性套细胞淋巴瘤(MCL)的成年患者。Pirtobrutinib与BTK野生型和C481突变体结合,从而抑制BTK激酶活性。
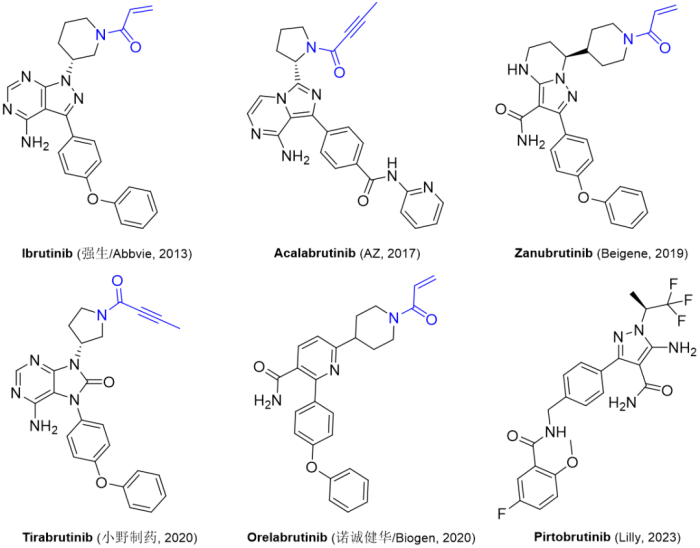
图2. 目前获批上市的BTK抑制剂。
1/27/2023: Orserdu (elacestrant)
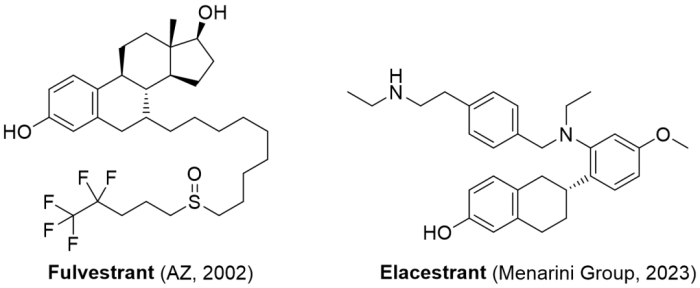
图3. 目前获批上市的选择性雌激素受体拮抗剂。
2/1/2023: Jesduvroq (daprodustat)
基本信息:Daprodustat是由GSK开发的口服HIF-PH(缺氧诱导因子-脯氨酰羟化酶)抑制剂,用于每日一次治疗接受透析至少四个月的成人因慢性肾病 (CKD) 引起的贫血。抑制HIF-PH可以稳定缺氧诱导因子,从而导致促红细胞生成素和其他参与纠正贫血的基因转录,类似于人体在高海拔地区发生的生理效应。CKD是一个日益增加的全球健康负担,影响着全世界7亿患者,据估计1/7的患者还患有贫血。CKD在美国影响着大约3900万人,其中大约600万人患有贫血。美国大约有810,000名终末期肾病 (ESRD) 患者。其中,有558,000名患者接受透析。
分子设计思路:脯氨酰羟化酶(prolyl hydroxylase, PH)属于含铁双加氧酶,PH需要2-氧代戊二酸 (2-OG) 作为底物和铁作为辅助因子,daprodustat(GSK-1278863)是以2-OG类似物NOG为原型,添加嘧啶三酮骨架得到。
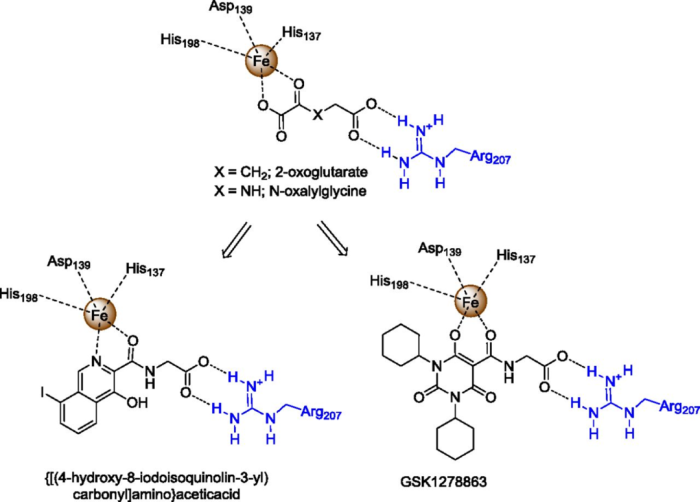
图4. HIF-PH抑制剂daprodustat(GSK-1278863)的分子结构。
2/17/2023: Filspari (sparsentan)
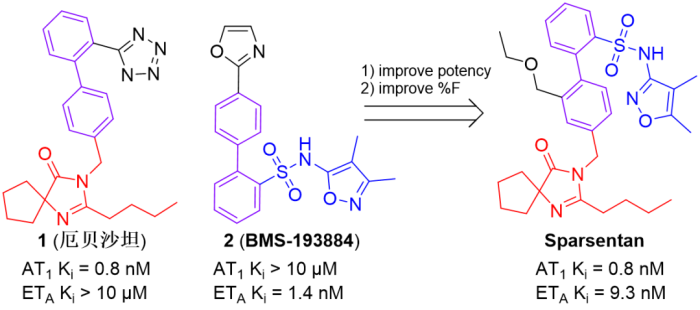 图5. Sparsentan的分子设计总结。
图5. Sparsentan的分子设计总结。
2/28/2023: Skyclarys (omaveloxolone)
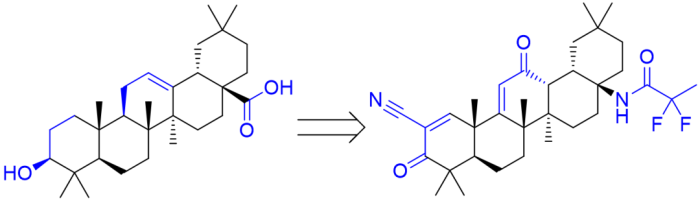
图6. 齐墩果酸引入亲电基团改造得到Omaveloxolone。
3/9/2023: Zavzpret (zavegepant)
分子设计思路:通过调研期刊和专利,总结已知CGRP受体拮抗剂共有的结构特征,得到hit,其Ki为0.55 nM,但该分子是CYP3A4的有效抑制剂且溶解性较差。对苯并噻吩侧链进行SAR,发现7-甲基吲唑在显著提高了活性且对CYP3A4有可接受的抑制。此外,在对喹唑啉酮的C-8位引入氟原子后,得到分子2(BMS- 694153)。不过2在水溶液中易被氧化,为此用缺电子sp2的次甲基替换对氧化敏感的苄基亚甲基,同时对哌啶-哌啶侧链进行简单的SAR改为N-甲基哌啶基-哌嗪(有两个可质子化的氮,提高水溶性),得到了BMS-742413(即上市药物zavegepant)。
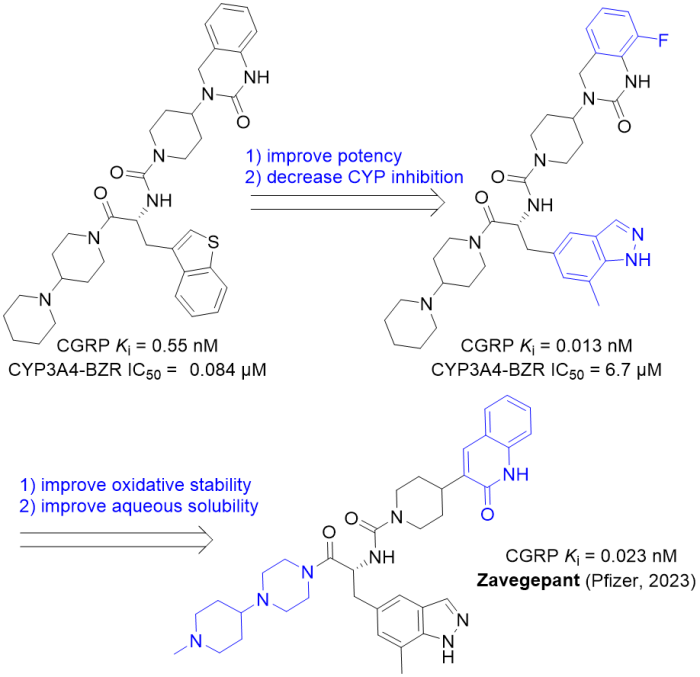 图7. Zavegepant的分子设计总结。
图7. Zavegepant的分子设计总结。
3/10/2023: Daybue (trofinetide)
基本信息:Trofinetide是由Acadia Pharmaceuticals公司(与澳洲Neuren Pharmaceuticals合作)开发的第一个用于治疗2岁及以上的Rett综合征患者的药物(口服溶液)。Rett综合征是一种复杂、罕见的神经发育障碍,通常由MECP2基因的基因突变引起,该基因的缺陷已被证明会导致突触通讯受损。Rett综合征的特点是在6-18个月之前有一段正常发育期,随后出现显著的发育倒退。
分子设计思路:Trofinetide是IGF-1(胰岛素样生长因子1)的氨基末端三肽(甘氨酸-脯氨酸-谷氨酸,GPE)的合成类似物(脯氨酸上多了一个甲基取代),提高了trofinetide对蛋白酶活性的代谢抗性和口服利用度,从而延长半衰期并做到口服给药。
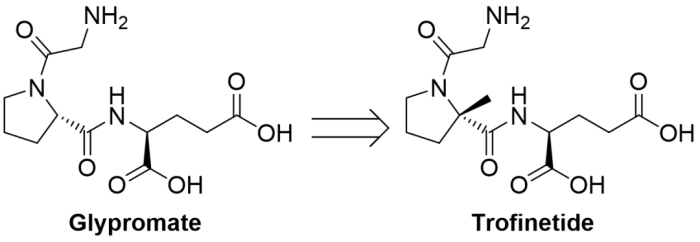
图8. Trofinetide的分子设计总结。
3/22/2023: Rezzayo (rezafungin)
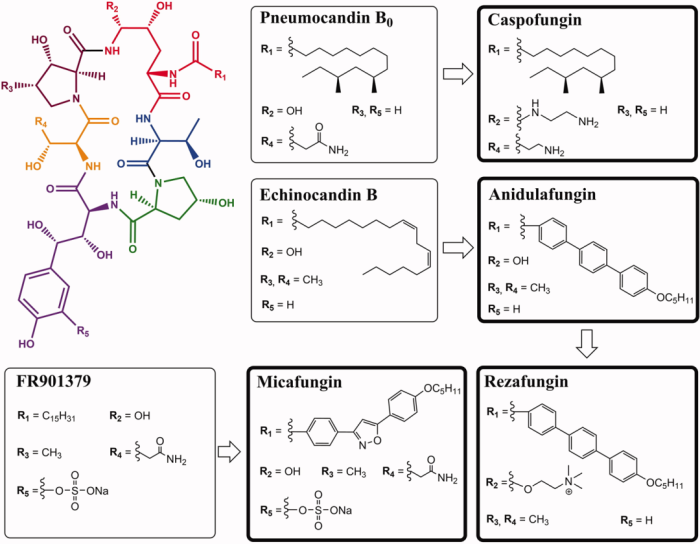
图9. 上市的棘白菌素类抗真菌药物的分子结构。
3/24/2023: Joenja (leniolisib)
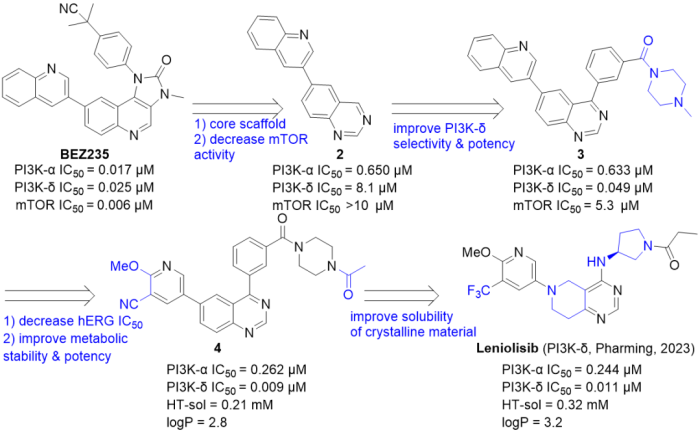 图10. Leniolisib的分子设计总结。
图10. Leniolisib的分子设计总结。
5/12/2023: Veozah (fezolinetant)
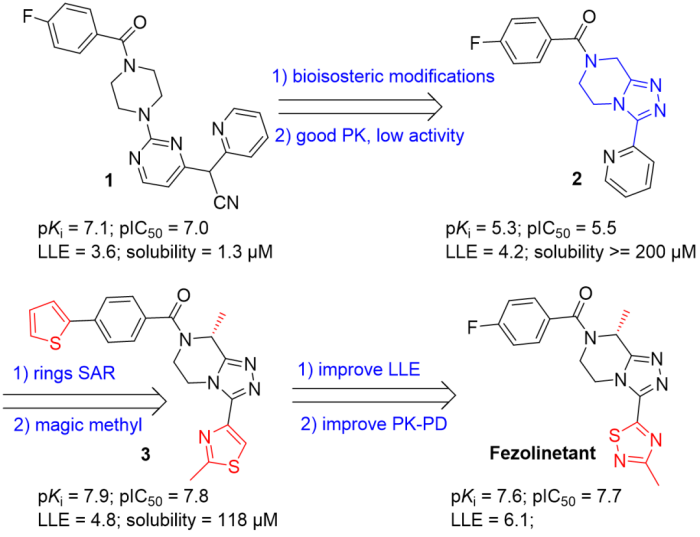 图11. Fezolinetant的药物设计。
图11. Fezolinetant的药物设计。
5/23/2023: Xacduro (sulbactam, durlobactam)
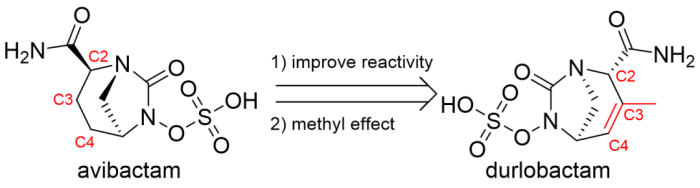 图12. Durlobactam的药物设计总结。
图12. Durlobactam的药物设计总结。
5/25/2023: Paxlovid (nirmatrelvir, ritonavir)
基本信息:Paxlovid是由Pfizer研发的新冠口服药物,由奈玛特韦(Nirmatrelvir)和利托那韦(ritonavir)组成,用于治疗有进展为重症风险的轻度至中度新冠患者。Nirmatrelvir是SARS-CoV-2-3CL蛋白酶(又称为主蛋白酶)的可逆共价抑制剂。
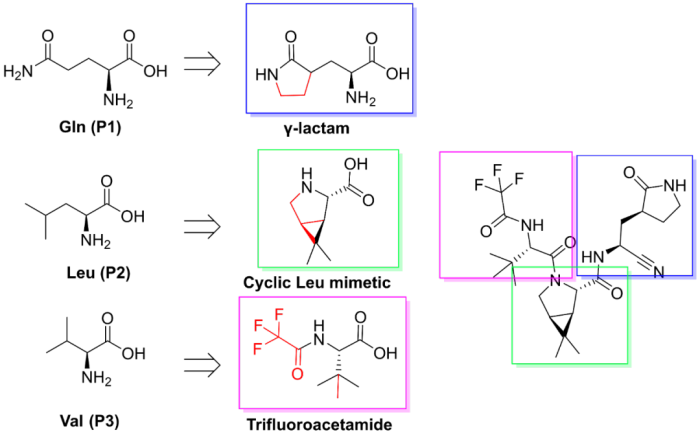
分子设计思路:Nirmatrelvir的P1和P2位点参照Gln和Leu改造为环状,借鉴了感冒药rupintrivir的γ-lactam片段和HCV NS3/4A蛋白酶抑制剂的氮杂双环。Nirmatrelvir的P3/P4位点则是基于改善溶解性、渗透性和口服利用度等成药性优化得到。最后通过PK研究发现加入利托那韦能改善代谢消除率,得到临床药物PAXLOVID™。更多关于Paxlovid的信息可以查看《从SARS到SARS-CoV-2,辉瑞新冠口服药物的分子设计》和《从感冒药到HCV药物,辉瑞新冠药物Paxlovid隐含的设计细节》。
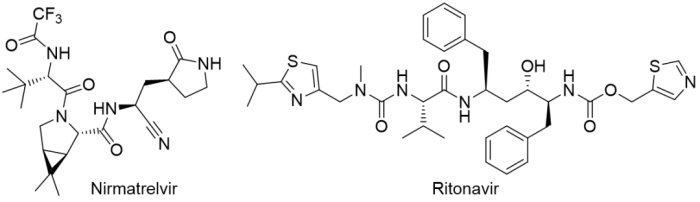 图13. 分别为Nirmatrelvir和ritonavir的分子结构。
图13. 分别为Nirmatrelvir和ritonavir的分子结构。
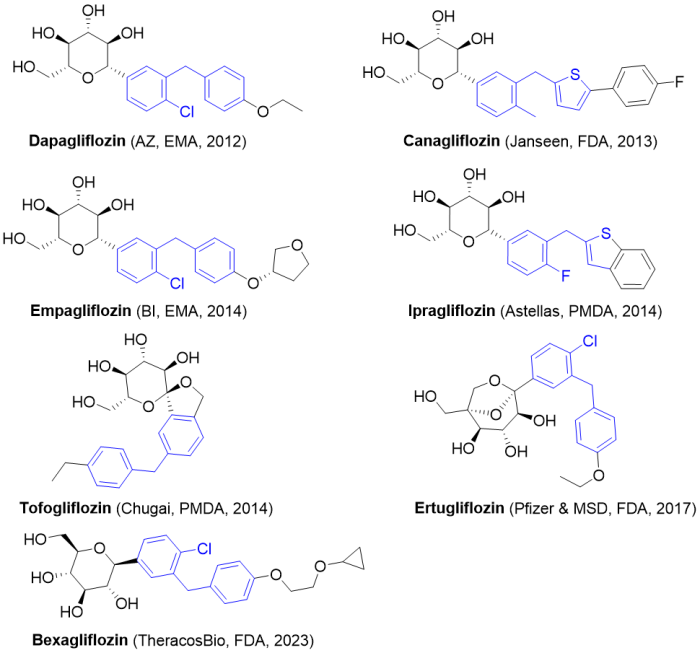
5/26/2023: Inpefa (sotagliflozin)
分子设计思路: 天然产物根皮苷(常见于苹果根皮中)是SGLT1(主要在肠道表达)和SGLT2的抑制剂,目前上市的多个SGLT2选择性抑制剂都是在根皮苷结构的基础上进行优化改造。Sotagliflozin的分子结构与AZ的达格列净(Dapagliflozin)非常相似,只是葡萄糖单元的5号位的羟甲基改为了甲硫基。
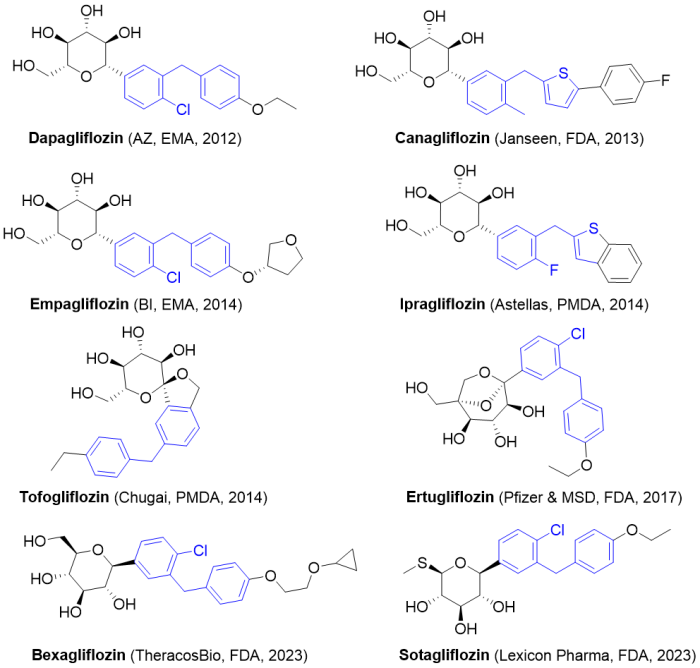 图15. 目前获批上市的部分SGLT2抑制剂。
图15. 目前获批上市的部分SGLT2抑制剂。
6/23/2023: Litfulo (ritlecitinib)
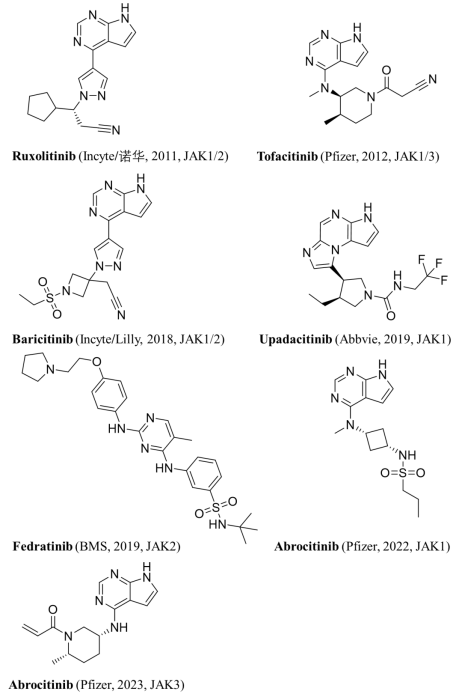 图16. 目前获FDA批准上市的JAK抑制剂(以FDA上市时间为准)。
图16. 目前获FDA批准上市的JAK抑制剂(以FDA上市时间为准)。
10/12/2023: Velsipity (etrasimod)
基本信息:Etrasimod是由Pfizer开发的1-磷酸鞘氨醇(S1P)受体调节剂,以高亲和力与S1P受体1、4和5结合,用于治疗成人中重度活动性溃疡性结肠炎(UC)。
Etrasimod部分地、可逆地阻断淋巴细胞从淋巴器官流出的能力,减少外周血中淋巴细胞的数量。UC是一种自身免疫性疾病,影响北美和欧洲约380万人,症状包括出血和慢性腹泻、腹痛和失禁等。
分子设计思路:首先通过内部HTS确定磺酰胺1是S1P1受体(EC50 = 6.7 nM)的有效激动剂。但芳基磺酰胺类似物在口服给药后不能有效降低小鼠的淋巴细胞计数。分别用吲唑和1,2,4-恶二唑取代苯环和磺酰胺,得到体内potency提高的分子2。随后,i)在吲唑环上引入酸性侧链,模拟S1P中发现的磷酸盐残基;ii)用二氢吲哚替换吲唑类环;iii)用氰基替换苯环的其中一个三氟甲氧基,发现二氢吲哚3在淋巴细胞降低实验中产生最大反应,将其作为lead。
3对心脏表达的S1P3亚型的选择性(S1P3的激动会导致啮齿类动物心动过缓)非常好,且半衰期相对上市药物Fingolimod缩短明显(长达6-9天,停药后外周淋巴细胞水平恢复正常需要1-2个月),符合最初的优化目标。遗憾的是静脉输注24小时后,在大鼠大脑和脑脊液中检测不到4。提高CNS渗透性(可通过直接作用于星形胶质细胞上表达的S1P1来减少神经炎症)是进一步优化的重点。用甲氧基替换1,2,4-恶二唑linker,并对苯环的取代基进行优化得到上市药物Etrasimod,其相对于4得到改善的CNS暴露,脑-血浆(B/P)比率为1.9。
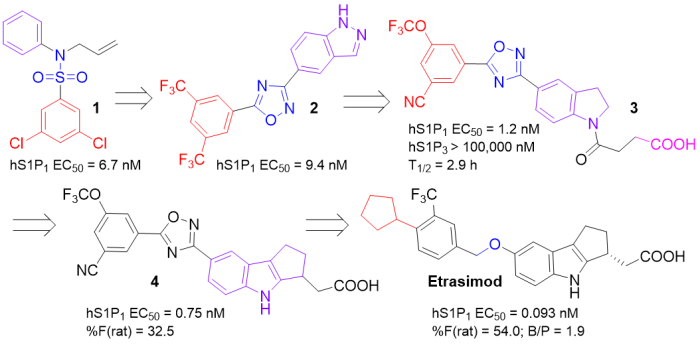
图17. Etrasimod的药物设计。
10/26/2023: Agamree (vamorolone)
基本信息:Vamorolone是由Santhera开发的是糖皮质激素受体的部分激动剂(皮质类固醇),用于治疗2岁及以上的杜氏肌营养不良症 (DMD) 患者。美国的DMD患者约有11,000-13,000人,目前的标准治疗是皮质类固醇(尽管其存在严重的不良事件),大约70%的患者同时接受皮质类固醇治疗。
DMD是最常见的肌营养不良症,是一种X连锁隐性遗传病(几乎只影响男性),其特征是DMD基因(人类基因组中最大的基因)发生突变,并且编码的肌营养不良蛋白丢失。DMD的特点是肌肉功能退化。患有这种罕见的致命疾病的患者会慢慢失去行走能力并出现呼吸衰竭,平均预期寿命低于30岁。
分子设计思路:Vamorolone显然是皮质醇(氢化可的松)和泼尼松龙(1,2-脱氢皮质醇)的衍生物,结构上少了羟基,多了双键和甲基。Vamorolone多出来的双键已被证明可以去除与糖皮质激素受体的分子接触位点,并导致“解离”特性,即反式激活活性相对丧失,但保留反式抑制活性。反式激活引发了GR介导的药物活性的许多安全问题,而反式抑制则提高了抗炎功效。
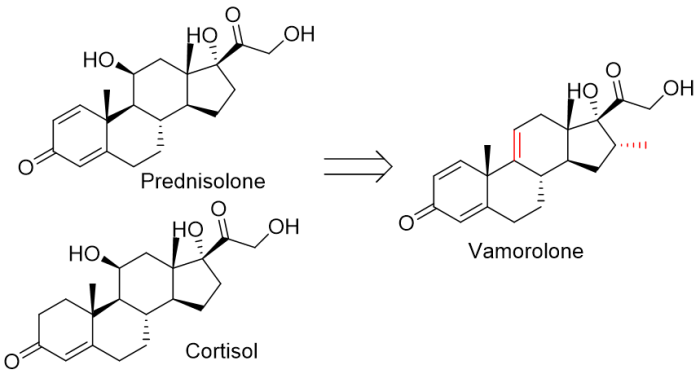
图18. Vamorolone的药物设计。
11/8/2023: Fruzaqla (fruquintinib)
基本信息: Fruquintinib(呋喹替尼)是由和黄医药开发(2023年1月以11.3亿美元授权给武田制药)的一款口服的高选择性、强效的VEGFR-1/2/3(IC50分别为33、35和0.5 nM,对其它激酶选择性较好)抑制剂,用于治疗既往接受过氟嘧啶、奥沙利铂和伊立替康治疗的转移性结直肠癌患者,包括既往接受过抗VEGF治疗和/或抗EGFR治疗(RAS野生型)的患者。
在美国,2023年将诊断出约153,000例新发CRC病例,占所有新发癌症病例的7.8%。大约70%的CRC患者无论是在诊断时还是治疗后都会出现转移性疾病。
分子设计思路:此前已有多款VEGFR抑制剂上市,其中市场表现最好的是Lenvatinib。如图3所示,呋喹替尼和Lenvatinib还是有不少相似之处。呋喹替尼主要改动的地方在杂环上多了一个N,以及用呋喃环替换了苯环上原有的极性侧链。
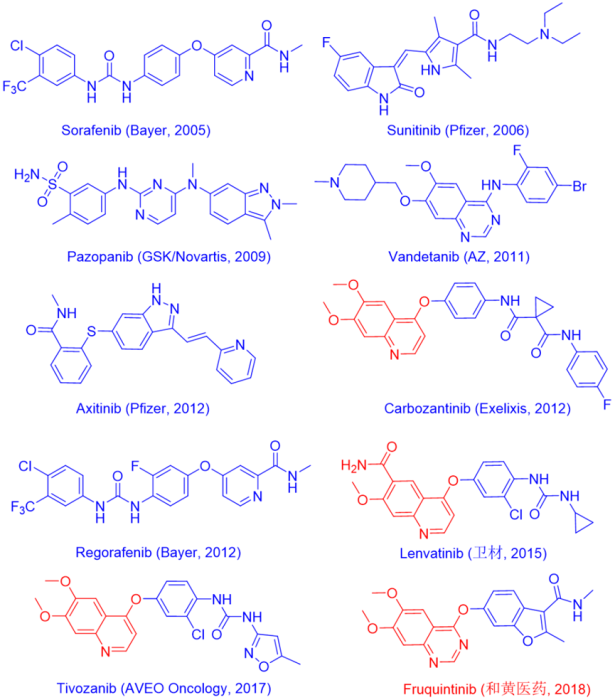
图19. 部分上市的VEGFR抑制剂。
11/15/2023: Defencath (taurolidine, heparin)
基本信息:Defencath是由CorMedix开发的是牛磺罗定(taurolidine,噻二嗪类抗菌剂)和肝素(抗凝剂)的组合,用于降低通过中心静脉导管(CVC)接受血液透析 (HD) 的成年肾衰竭患者导管相关血流感染 (CRBSI) 的发生率。CRBSI在CVC患者中很常见。大约80%开始血液透析的患者将插入CVC以建立血管通路。每年使用CVC等血管内装置的患者会发生约250,000例CRBSI,导致约1/4的感染患者死亡。血液透析中的 CRBSI由多种病原体引起,其中许多病原体具有抗生素耐药性。
分子设计思路:没找到牛磺罗定的药物发现文章。牛磺罗定源自内源性氨基酸衍生物牛磺酸。牛磺罗定在体内代谢为牛磺酸酰胺,最终生成牛磺酸和水,释放羟甲基,与细菌细胞壁中的胞壁蛋白以及内毒素和外毒素的氨基和羟基发生化学反应。该反应使细菌细胞壁的内毒素以及复杂的多糖和脂多糖成分变性,并使敏感的外毒素失活。
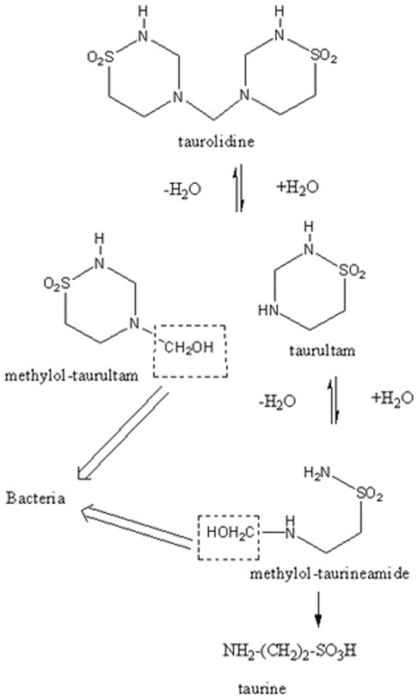
来源:PLoS ONE 2010, 5(1): e8927.
11/15/2023: Augtyro (repotrectinib)
基本信息:Repotrectinib是由BMS开发(2022年41亿美元收购Turning Point获得)的酪氨酸蛋白激酶(ROS1)抑制剂,用于治疗局部晚期或转移性ROS1阳性NSCLC成年患者。Repotrectinib是原癌基因ROS1和TRKA、TRKB和TRKC的抑制剂。包含ROS1结构域的融合蛋白可以通过下游信号通路的过度激活来驱动致瘤潜力,从而导致不受限制的细胞增殖。
ROS1融合发生在约1-2%的NSCLC患者中。ROS1阳性肿瘤患者的平均年龄为50岁,往往比普通肺癌患者更年轻,其中女性多于男性,并且可能几乎没有吸烟史。ROS1阳性肺癌往往具有侵袭性,通常可扩散至大脑。
分子设计思路:由于TRK激酶结构域的二次突变,总是会产生获得性耐药。TRKA中最常见的突变为G595R和G667C,分子模型表明, G595R突变引入了larotrectinib羟基吡咯烷基团的空间位阻(图5)。同样, G667C突变增加了larotrectinib的二氟苯基的空间位阻。为了克服上述耐药突变,开发了两种大环化合物LOXO-195和TPX-0005(即Repotrectinib),与larotrectinib的骨架相比,引入更大的构象刚性,并避免与R595和C667的空间clash。
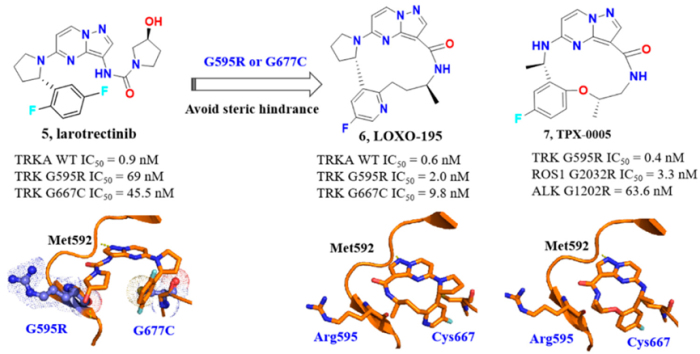
来源:J. Med. Chem. 2020, 63, 10726–10741.
11/16/2023: Truqap (capivasertib)
基本信息:Capivasertib是由阿斯利康开发的丝氨酸/苏氨酸激酶AKT(AKT1-3)的ATP竞争性抑制剂,与氟维司群联合用于治疗患有激素受体(HR)阳性、HER2阴性、患有一种或多种PIK3CA/AKT1/PTEN改变的局部晚期或转移性乳腺癌成年患者。PI3K-Akt-mTOR通路是人类癌症中最常失调的信号通路之一,已被证明可以介导对细胞毒性、抗激素和靶向治疗的耐药性。Akt是该信号通路中的关键节点。
2020年,全球有超过200万患者被诊断患有乳腺癌,近685,000 死亡。HR阳性乳腺癌是最常见的乳腺癌亚型,超过65%的肿瘤是HR阳性且HER2低或阴性。PIK3CA、AKT1和PTEN突变影响高达50%的晚期HR阳性乳腺癌患者。
分子设计思路:高通量的fragment虚拟筛选将氮杂吲哚1鉴定为PKB(即Akt)的ATP竞争性配体。该片段通过SBDD优化为6-苯基嘌呤2。用哌啶取代2中的苯基并引入4-氨基得到3(碱性胺与Glu和Asn分别形成盐桥和氢键)。为了与P-loop的亲脂口袋相互作用,引入芳香族基团,得到氯苯基取代的分子4。当用4-氯苄基替换4-氯苯基,得到的分子5对PKA的选择性提高至约30倍。但5具有高体内清除率和低口服生物利用度,需要进一步优化。尝试了不同位置的SAR,发现通过简单地在5的4-氨基哌啶引入4-酰胺(调整了pKa/溶解度),即可得到PK显著提高的分子6。
然而,分子6对ROCK2激酶的选择性仅为5倍(ROCK2抑制剂已被证明可显著降低血压并导致心率和心肌收缩力增加),且对hERG的IC50达到5μM。突破点在于苄基的α-碳取代,首先用碱性侧链取代,发现携带碱性基团的侧链会导致高清除率和低口服生物利用度;随后引入的中性尤其是含羟基(改善hERG抑制活性的常见策略)的侧链则显著提高了对ROCK2和hERG的选择性,最终得到上市药物Capivasertib。
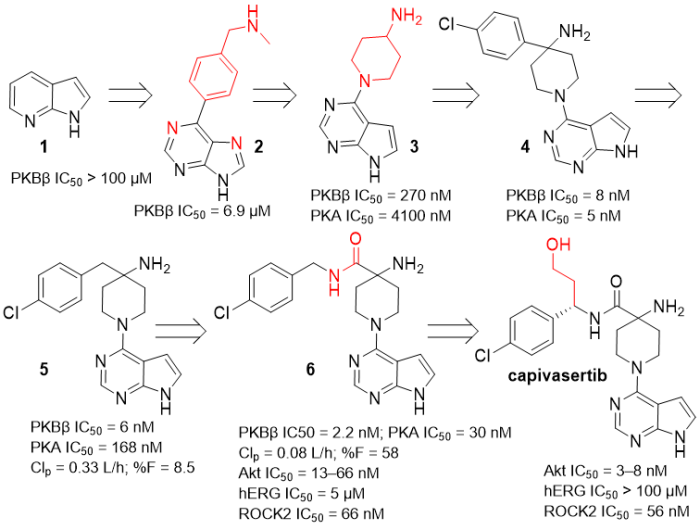
图22. Capivasertib的药物设计。
11/27/2023: Ogsiveo (nirogacestat)
基本信息:nirogacestat是由SpringWorks Therapeutics开发的口服γ分泌酶抑制剂,用于治疗需要全身治疗的进展性硬纤维瘤成年患者。硬纤维瘤是局部侵袭性和侵袭性软组织肿瘤,可导致严重的发病率。此外,当重要结构受到影响时,硬纤维瘤可能会危及生命。虽然硬纤维瘤不会转移,但现有的超适应症全身治疗通常难以治愈,并且手术切除后复发率高达77%。
分子设计思路:没找到nirogacestat的药物发现文章。查阅文献nirogacestat应该是参考二肽类似物DAPT(Aβ IC50 = 20 nM)优化而来。
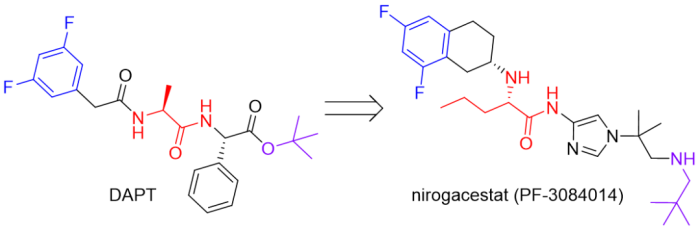
图23.nirogacestat的药物设计。
12/5/2023: Fabhalta (iptacopan)
基本信息:旁路途径(AP)是补体激活的放大环,是各种人类疾病的促成因素,包括年龄相关性黄斑变性(AMD),阵发性睡眠性血红蛋白尿(PNH)等。AP的补体系统成分包括蛋白酶因子B (FB)和蛋白酶因子D (FD)等。FB是一种类胰蛋白酶丝氨酸蛋白酶,主要在肝脏和巨噬细胞产生,以潜伏形式在人体血液中循环。通过抑制FB可以阻断AP放大循环,用于治疗多种补体介导的疾病。
Iptacopan是由诺华研发的FB抑制剂,用于治疗PNH。Iptacopan与补体旁路途径的FB结合,调节C3的裂解、下游效应子的产生以及末端途径的扩增。在PNH中,血管内溶血 (IVH) 由下游膜攻击复合物 (MAC) 介导,而血管外溶血 (EVH) 由C3b调理作用促进。Iptacopan在补体级联的替代途径中发挥近端作用,控制C3b介导的EVH和末端补体介导的IVH。
分子设计思路:通过高通量筛选鉴定出氨基咪唑啉1(IC50 = 6.6 μM,SPR Kd = 10 μM)片段。考虑到萘环骨架不容易进行SAR,且萘基的非极性性质可能会使与相对极性的S1口袋很难产生positive的相互作用,用相对极性的吲哚环取代萘环。同时,用不同碱度的胺来替换咪唑啉,发现哌啶衍生物18的IC50为50 μM。18的结构简单(MW = 242.4),加上哌啶的C-2和C-4方便衍生化,从而提供新的SAR位点:1)在哌啶的2位引入苯基得到化合物19,其共晶结构显示哌啶的C-4位能够生长到S3口袋中;2)在哌啶的C-4位引入-OMe得到的21可以显著提高效力,还表现出α2c受体结合亲和力的显著降低 (IC50 = 11 μM)。值得一提的是,当21的苯基换为羟基,得到的哌啶29相比21效力提高了2倍。
29与FB的共晶揭示了羟基和结晶缓冲液中的硫酸根离子之间的相互作用,与Arg192、Val218和Asn220B形成了复杂的氢键网络。为了进一步提高potency,尝试通过在哌啶的C-2位置引入极性官能团来模拟这种相互作用。重新评估了29和19与FB结合的晶体结构,发现19中苯环的对位位置完美,可以将取代基放入硫酸盐占据的空间中。在苯环对位连上羧酸,得到的35相比29的potency提高了近100倍。此外,酸的添加消除了hERG和肾上腺素能受体活性,但35在不同物种间存在功能活性差异。猜测填充S3口袋应该会增强对抗人类酶的效力,也可能导致小鼠效力随之增加。最后,在哌啶的C-4处用乙氧基替换甲氧基能更深地扩展到S3口袋,用吲哚C-5甲氧基替换甲基提供了更大程度的眼部AP抑制,最终得到上市药物iptacopan。
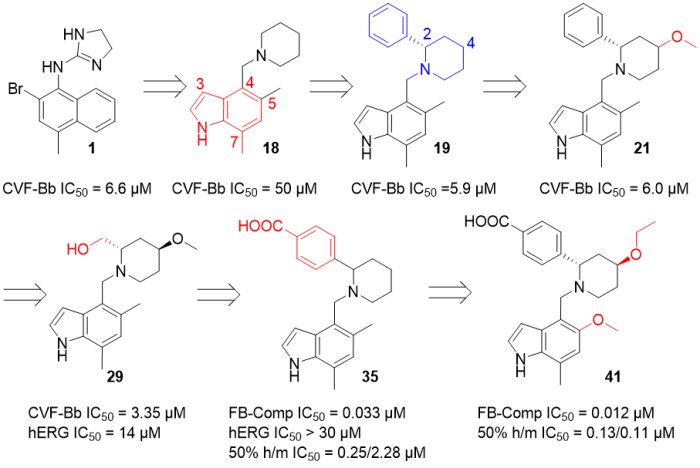
图24. Iptacopan的药物设计。
撰稿人 | 智药邦
责任编辑 | 邵丽竹
审核人 | 何发
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多