mRNA疫苗的发展历程与作用机制
在随后的研究中发现,mRNA可以发挥类似疫苗的作用以达到治疗的目的。进入21世纪后,mRNA的合成技术、修饰技术和递送技术进一步发展,相比传统疫苗,mRNA疫苗具有更安全、效率更高和免疫原性更好的优势,且不会插入基因突变,可以被正常细胞降解,还可以通过序列修饰和递送载体改变其半衰期,同时,还容易大规模生产。

通过将含有编码抗原蛋白的mRNA导入人体,可以跳过复制、转录过程,直接进行翻译,形成相应的抗原蛋白,从而诱导机体产生特异性免疫应答,达到预防免疫的作用。
以新型冠状病毒mRNA疫苗作用原理为例:
刺突糖蛋白(S蛋白)是新冠病毒入侵人体的关键蛋白,这种蛋白可以识别人类呼吸道上皮细胞上的ACE2蛋白,并与之相互作用,从而打开新型冠状病毒入侵人体的大门。简而言之:ACE2是新冠病毒S蛋白的受体。
新型冠状病毒mRNA疫苗是将编码新冠病毒刺突糖蛋白(S蛋白,抗原蛋白)的mRNA直接注射进入体内,能够在人体内合成S蛋白,通过模拟病毒感染刺激机体产生抗体和记忆细胞。

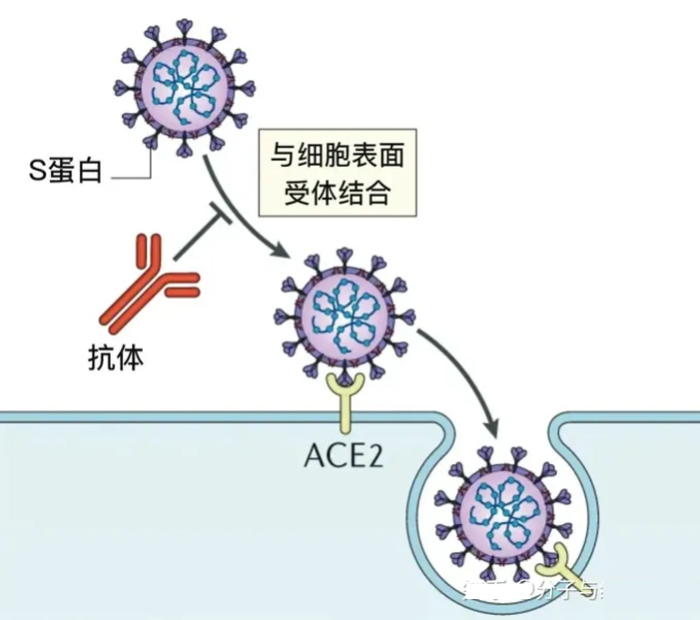

目前应用的mRNA疫苗主要有两种形式:
①基于mRNA的传统mRNA疫苗(非复制型mRNA疫苗)
定义:是在体外转录好的一段编码抗原蛋白的完整mRNA,上游和下游分别包含5′帽子结构和 3′poly(A)尾,只编码目标抗原。
优点:结构简单、RNA序列短、不编码其他蛋白
缺点:在体内半衰期短、抗原表达量低,需要较高的量才能诱发有效的免疫应答。
②自扩增型(self-amplifying, SAM)mRNA疫苗(又称复制子)
定义:是通过基因工程改造的 mRNA病毒基因组,其中编码RNA复制机制的基因是完整的,用编码抗原蛋白的mRNA代替了原病毒的结构蛋白编码基因,可以在体内实现自我扩增,很少的量就可以诱发有效的免疫应答。

1.RNA病毒感染性疾病
mRNA疫苗可用于传染性强、危害性大的RNA病毒感染性疾病的预防,如新冠病毒感染,目前已经在预防新冠病毒感染方面起到重要作用。
2.DNA病毒感染性疾病
mRNA疫苗可用于巨细胞病毒等DNA病毒感染性疾病的预防。
3.细菌感染性疾病
mRNA疫苗还可用于链球菌和结核杆菌等细菌感染性疾病。

【优点】:
①抗原选择范围广;
②外源mRNA可激活模式识别受体(pattern recognition receptor,PRR),刺激非特异性免疫反应,进而激发T、B 细胞免疫反应,具有“自我佐剂(self-adjuvant)”的特点,表现更强的免疫原性;
③与质粒DNA 疫苗相比不进入细胞核内部,只在细胞质内表达抗原,不存在整合人体基因上的风险;
④不依赖细胞培养技术,可快速构建疫苗,外源因子传播风险较低。
【缺点】:
①体内稳定性差;
②低免疫原性;
③裸mRNA 无法被有效递呈至细胞内并成功从溶酶体逃逸,体内体液和细胞免疫较低,因此需要脂质体增强细胞摄取并改善向细胞质转译机制的传递,从而提高mRNA 疫苗的作用。
【难点】:
①mRNA进入体内后易被核糖核酸酶降解,递送技术相对复杂;
②如何提高mRNA稳定性及翻译效率;
③调节免疫原性;
④如何开发高效无毒的递送载体。
1. 有研究显示,mRNA 可激活Ⅰ型干扰素介导的先天性免疫反应,引起真核翻译起始因子2(eukaryotic initiation factor 2,eiF2)的磷酸化,导致蛋白翻译减缓并最终抑制。
2. 研究表明,一种用于预防狂犬病的mRNA 疫苗和一种编码H7N9 或H10N8 病毒血凝素的mRNA 流感疫苗在人体中的中和抗体滴度低于动物模型,有效性与动物模型差异较大。
3. 安全性方面,临床试验结果显示多个mRNA 疫苗可引起受试者轻至中度局部或全身反应,因此需要在合适的动物模型上评估mRNA 疫苗的分布,所编码的抗原在远端器官的表达及潜在的安全风险,包括对局部和全身的影响。产品安全性风险可能来源于mRNA、脂质递送系统两个方面,非临床研究中需关注脂质的毒性及在人体内诱导自身反应性抗体等。

【注意】:mRNA疫苗具有高效、安全、成本较低等优点,但在身体中的表达不够稳定,可能会引起一些局部反应或全身反应等副作用,如果身体处于感冒或者发热等情况下,是不可以注射疫苗的,否则会产生不良反应。

根据疫苗生产技术和工艺不同,大致可以将疫苗分为三代。
第一代疫苗:
有减毒活疫苗和灭活疫苗。
减毒活疫苗:是通过自然选取或人工培育致病力低下或不致病的病毒株,使原本致病的病毒或细菌毒力下降到不能致病,但可刺激人体产生免疫应答的状态,以获得对特定疾病的免疫力。
常见的减毒活疫苗包括卡介苗、脊灰减毒活疫苗等,其特点是免疫效果好、作用时间长,缺点是可能存在“毒力返祖”的现象,并且对储存和运输要求较高,需要全程冷链运输保存;
灭活疫苗:又被称为“死疫苗”,没有致病力,可让人产生相应的疾病免疫力,
常见的灭活疫苗包括百白破疫苗、新冠灭活疫苗等,其特点是安全性较好,不会出现“毒力返祖”现象,对储存和运输条件的要求不高,节省了疫苗成本,缺点是它引起机体产生的免疫力持续时间较短,通常需要多剂次接种。
第二代疫苗:
有亚单位疫苗和重组基因疫苗。
亚单位疫苗:是通过蛋白质水解,提取、筛选具有免疫活性蛋白质片段制成疫苗,亚单位疫苗相较全病毒疫苗来说安全性和稳定性都更好,缺点是降低了疫苗的免疫原性,可能发生抗原变性导致产生的抗体失效;
重组基因疫苗:是将病毒内能够诱导免疫反应发生的基因片段植入到工具细胞中,规模化的生产我们所需要的蛋白质,再收集、纯化这些蛋白质后就可以制成重组基因疫苗,如乙肝疫苗等。
第三代疫苗:
有重组病毒载体疫苗和核酸疫苗。
重组病毒载体疫苗:是将病毒中能够诱导合成具有免疫原型的蛋白质或亚单位的基因片段嫁接到没有致病性或致病性极低的病毒中,该载体病毒携基因片段进入人体,就能够诱导免疫反应的发生;
核酸疫苗:又称基因疫苗,包括DNA疫苗和mRNA疫苗。核酸疫苗是将决定病毒或细菌免疫原性的遗传物质直接导入人体,依靠人体自身细胞来产生抗原,并激活免疫系统产生免疫应答,两者的区别是DNA是先转录成mRNA再进行抗原蛋白质的合成,mRNA则是直接合成。目前在研的新冠疫苗大多属于mRNA疫苗。
mRNA疫苗作为目前最新的疫苗生产技术,优势明显,不用依赖细胞扩增,制备工艺简单、研发周期短,疫苗产能得到了大大的提升,缺点在于,由于mRNA极易降解,需借助特殊载体送达体内,并且对运输和储存要求也非常严格。

1961年,发现mRNA
1965年,制造出首个脂质体
1969年,首次在实验室分离mRNA合成蛋白
1971年,脂质体用于递送药物
1974年,脂质体用于递送疫苗
1978年,首次将脂质体包裹的mRNA递送至细胞
1984年,实验室合成mRNA
1989年,将合成mRNA包裹在阳离子脂质(带正电的脂质结构)中递送到人体细胞、青蛙胚胎
1990年,将脂质体包裹的mRNA递送到小鼠体内
1992年,测试mRNA在大鼠中的疗法
1993年,测试首个mRNA疫苗,用于预防小鼠流感
1995年,mRNA作为癌症疫苗在小鼠中测试
1997年,首个mRNA公司成立,Merix Biosciences,后更名为Argos,现在为Colmmune)
2000年,CureVac公司成立
2001年,首次报道含四种成分的脂质纳米粒(当时用于递送DNA)
2005年,发现修饰的RNA能逃逸免疫检测,发现量产脂质纳米粒的技术
2008年,BioNTech公司成立,诺华和Shire成立mRNA部门
2010年,Moderna公司成立
2012年,小鼠中测试首个脂质纳米粒包裹的mRNA疫苗
2013年,首次对预防传染病(狂犬病)的mRNA疫苗开展临床试验
2015年,首次对脂质纳米粒包裹的mRNA疫苗(流感)开展临床试验
2018年,首个使用脂质纳米粒的药物(patisiran)获批
2020年,新冠mRNA疫苗紧急授权使用
数据来源:自U. Şahin et al. Nature Rev. Drug Discov. 13, 759–780 (2014)和X. Hou et al. Nature Rev. Mater. https://doi.org/gmjsn5 (2021).

mRNA疫苗和灭活疫苗是临床常见的两种疫苗,对于临床疾病预防意义重大。
二者具有以下区别:
一是制作工艺有区别,mRNA是DNA翻译成蛋白质的中间产物,mRNA疫苗是用各类病原微生物的mRNA制作,而灭活疫苗是经化学或物理等方法灭活的病原微生物制成的疫苗;
二是作用原理有区别,mRNA疫苗是一种模拟天然mRNA结构与功能,将感兴趣基因导入体内,通过诱导机体免疫应答,实现预防传染病或抗肿瘤等目的的重组疫苗,而灭活疫苗虽然在机体内失去繁殖能力,但能刺激机体产生良好免疫反应,如产生特异的抗体或致敏T淋巴细胞,进而达到预防疾病的作用;
三是效果评价有区别,病毒灭活疫苗工艺简单,但是免疫效价相对较低,mRNA疫苗制作速度快,但容易降解。

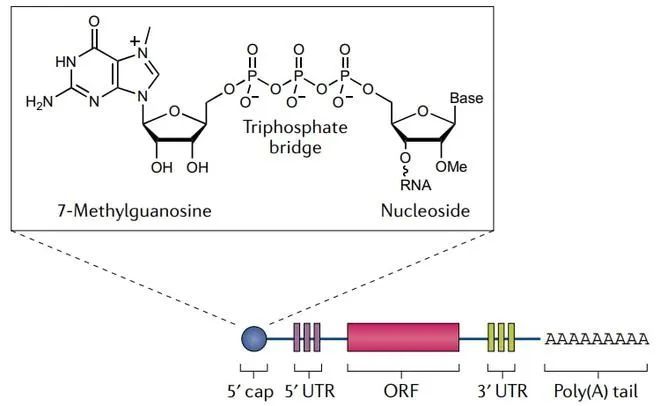
1.帽子结构:是真核生物在转录后修饰过程产生的位于5’端的特殊结构。5’端帽包含一个通过三磷酸桥连接到mRNA 5’端的7-甲基鸟苷。帽子结构包裹住mRNA的5’端,使其免于核酸外切酶的降解;还可促使mRAN环化,并募集核糖体启动翻译进程。
2.非翻译区(UTR):包括5’UTR和3’UTR两个部分,可调节mRNA的翻译和半衰期等。5’UTR在帽子结构和编码区ORF之间,其长度和二级结构都会对翻译起始阶段的效率产生影响。3’UTR位于编码区ORF下游,在终止密码子之前。真核生物mRNA 的 3’UTR 是其重要功能元件,在翻译调控和mRNA 定位等方面起着不可忽视的作用。
3.编码区ORF:由起始密码子开始,到终止密码子结束。每三个碱基构成一个密码子,编码一个氨基酸。编码区若存在过多的二级结构和稀有密码子则会降低翻译效率。因此,“mRNA三巨头”之一的CureVac将ORF中第三个位置的A或U替换为更常见的G或C,并将这一优化策略用于其COVID-19候选疫苗CVnCoV中。但有些时候,翻译较慢的稀有密码子对保证蛋白质准确折叠很有必要,因此使用这种替换方法还须谨慎。
4.3’UTR尾部:是一段Poly A序列。与帽子结构类似,Poly(A)尾也能起到保护mRNA防止被核酸外切酶降解的作用。尾部结构也参与到翻译及其调控过程中,可与多聚腺苷酸结合蛋白(PABP)结合形成环状复合物,参与翻译的起始过程。
1. Katalin Karikó等科学家发现,在mRNA中掺入修饰核苷(假尿苷、N6-甲基腺苷、5-甲基胞苷)后可大大提高mRNA的翻译量。
2. 修饰核苷对在抑制RNA降解、提高翻译效率和调节半衰期等方面也发挥着重要作用。
3. 未修饰的mRNA(如病毒RNA)会被模式识别受体识别,从而产生免疫刺激。
辉瑞/BioNTech的mRNA疫苗BNT162b2和Moderna的mRNA-1273都利用了核苷修饰技术,用假尿苷替换尿苷。
CureVac的mRNA疫苗CVnCoV使用未经修饰的天然mRNA,通过其RNActive 平台改变mRNA序列并优化密码子,以提高mRNA的稳定性和免疫原性。


mRNA疫苗的生产可分为三大阶段,一是DNA原液制备,二是mRNA原液的制备,三是利用脂质微粒进行包封。
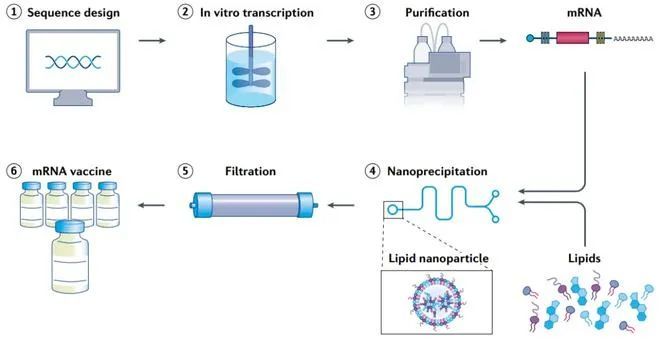
以辉瑞/BioNTech新冠疫苗BNT162b2的生产流程为例:
第一步:DNA质粒制备
原液制备开始于质粒构建。通常使用的DNA质粒为环状质粒,质粒上含有设计好的序列模块,其中包括刺突蛋白编码。利用电流打破细胞膜,并将环状DNA质粒引入大肠杆菌。此后,大肠杆菌被储藏于含有大量营养物质的溶液中进行繁殖扩增。
大肠杆菌每20分钟分裂一次,4天内可以得到数万亿DNA质粒。提取并纯化DNA质粒,过滤溶液,去除细菌及其他物质。利用酶将纯化后的环状DNA质粒切割为链状。
将所得溶液分装冷藏,通过质量控制环节,并运送至下一阶段的生产加工场所。此阶段生产过程耗时10天,若将质控和运输环节纳入考虑则共耗时约17天。
第二步:体外转录
目的是将DNA链转化为mRNA。上一步制备得到的DNA链与酶和核苷酸混合在10加仑容量的容器中,RNA聚合酶(RNA polymerase)会将DNA转录为mRNA。这一步骤被称为体外转录(IVT)。
得到mRNA后,DNA以及其他物质将被滤除,mRNA被装进购物袋大小的塑料包装中,每袋含有约500万到1000万剂次的mRNA原料。经过经过质量控制和运输到达下一生产环节,这一阶段的生产耗时约4天,质量控制和运输耗时12天,共16天。
第三步:递送系统装载
目的是将mRNA包裹进脂质载体(LNP)中。脂质悬浮于酒精溶液中,与mRNA接触并将其包裹,两种物质通过相反电荷相吸引。之后,原液经过切向流过滤(TFF)滤除溶液中多余的脂质、酒精等杂质,并制成最终的mRNA疫苗溶液。此生产过程耗时约12天。
第四步:灌装检验
在上述三个生产阶段都完成后,mRNA疫苗原液已完成,只待灌装分发。
(辉瑞一管容器中包含6剂次的疫苗原液量。辉瑞位于密歇根州的工厂能够在2天内完成100万剂次的灌装。随后,经过2周的纯净度检测及其他安全性检测,疫苗便能销往世界各地。此过程共花费约19天。以上所有生产灌装环节耗时约64天。)

1. mRNA序列结构决定抗原蛋白结构、免疫原性及稳定性
疫苗产生的抗原蛋白的序列以及稳定性决定了其激活的特异免疫的精确性和活性。而抗原蛋白的序列和结构则由mRNA序列影响和控制。因此,mRNA序列决定了疫苗的质量,也是mRNA疫苗厂商的核心竞争力之一。
生物学“中心法则”:DNA转录为mRNA转译为蛋白质。事实上,mRNA中只有部分片段会成为组成蛋白质的序列,其余部分则控制调节蛋白质的转译效率和结构。mRNA序列中,最终被转译成为蛋白质组成序列的部分被称为编码区域(translatedregion),编码区域前后分别有一个非转译区(untranslated region,UTR)。
编码区域决定了蛋白质中的氨基酸序列。编码区域中三个碱基为一组,称为密码子(codon)。密码子经转译成为特定的氨基酸,氨基酸串联后形成肽链,结构化后成为蛋白质。部分密码子组合会转译成同一种氨基酸(同义密码子Synonymouscodon),但在不同生物族群中,会存在对某一密码子组的偏好,此密码子组合的免疫原性较小,不易遭到酶类的攻击,而它的同义密码子则可能引起过敏反应并且导致质量下降。例如CAA、CAG均对应谷氨酰胺(Gln),但人类基因组中使用CAG更频繁,频率达到73%。因此CAA相较而言更有可能触发免疫反应。因此,mRNA疫苗需要选择最接近人源性的同义密码子,规避可能引起过敏反应的组合以保证安全性和转译质量。非转译区则调控mRNA和蛋白质的稳定性以及表达效率。
mRNA序列是疫苗研发中的重点,也是行业内竞争的核心。除了含有抗原蛋白编码外,序列其他部分也直接影响mRNA疫苗的质量。
编码前后的非转译区(UTR)负责调控转译以及蛋白表达,对mRNA的转译效率、半衰期、最高表达水平等数值有影响。转译效率可用来推断相同单位mRNA疫苗产生抗原的速度、半衰期用以推断mRNA能够产生抗原蛋白的时间、表达累计最高水平用以推断每单位mRNA在某一时刻能产生的抗原量的最高水平。同时,UTR中的GC水平、U水平均会影响mRNA的免疫原性,对疫苗的安全性和能否正常产生抗原造成影响。UTR需要在DNA质粒建立时包含在序列中,属于序列设计的一部分。
3’端多聚A尾(poly-A tail;A指腺苷,4种基础核苷酸的一种)位于mRNA尾部3’端,长度约100-250单位。多聚A尾能够提高mRNA的稳定性和转译效率。腺苷(A)能够降低核糖核酸酶(RNases)的效率,以此减缓mRNA的降解速度。多聚A尾可在建立DNA质粒时直接包含于编码中,也可在DNA转录为mRNA后通过聚合酶(poly-A polymerase,PAP)添加于mRNA尾部。
同时,位于mRNA最前端的5’帽结构对于降低mRNA免疫原性,增强稳定性和翻译效率有正面影响。5’帽结构是一个位于mRNA序列之前的反向7-甲基鸟苷(m7G)。在体外转录(IVT)过程中,mRNA5’端会含有三磷酸盐部分,具有很强的免疫原性。若不去除这一部分,当mRNA被递送进入人体细胞后,mRNA会在细胞质中引起IFN-1免疫反应,无法在人体内正常转译产生蛋白抗原。
加帽过程可在DNA转录为mRNA的生产过程中或过程后进行,利用抗反向帽类似物(ARCA)将5’帽结构按正确方向固定在mRNA的5’端。但是此过程不能保证三磷酸盐完全被去除,所以仍旧存在引起细胞内免疫反应的风险,因此加帽工艺对疫苗的安全性有直接影响。
2. 递送系统是目前产能扩张的瓶颈
作为mRNA疫苗的传递介质的一大难点是mRNA以内含体的形式进入细胞质后,需要打破内含体包膜,释放mRNA。目前LNP供应商较少,且其中专利纠纷因此成为mRNA新冠疫苗快速放量的一大掣肘。
目前辉瑞/BioNTech以及Moderna所使用的LNP专利都来源于一家名为Arbutus的加拿大生物公司。Arbutus将其部分LNP专利租借给另外两家公司Acuitas和Genevant,而这两家公司分别为Moderna和BioNTech的LNP专利来源。
3.LNP专利的不可替代
在其他载体均有明显劣势的情况下,LNP是mRNA疫苗递送的最佳选择之一。因此,各厂商对LNP技术的掌握以及是否拥有相关专利成为了业内竞争中极其重要的一环。厂商若无相关专利,则在竞争中会处于被“卡脖子”的尴尬境地。因此,LNP技术专利决定了mRNA疫苗企业在业内竞争中的地位。
撰稿人 | 生物制品圈
责任编辑 | 胡静
审核人 | 何发
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多