中国GMP|2025年新版无菌药品附录(征求意见稿)到底有哪些变化?

硬件差距:
PUBSIT
延展层流
压差/温湿度报警
无菌区硬件条件等
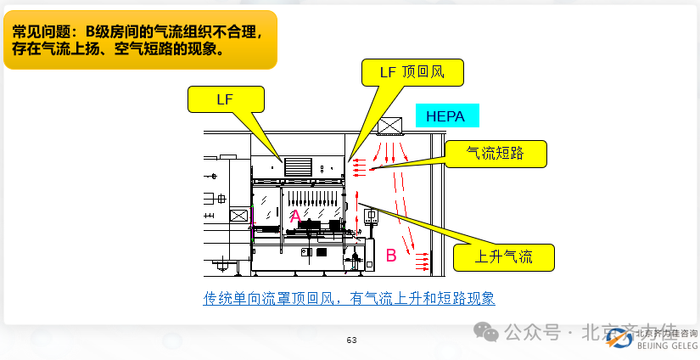
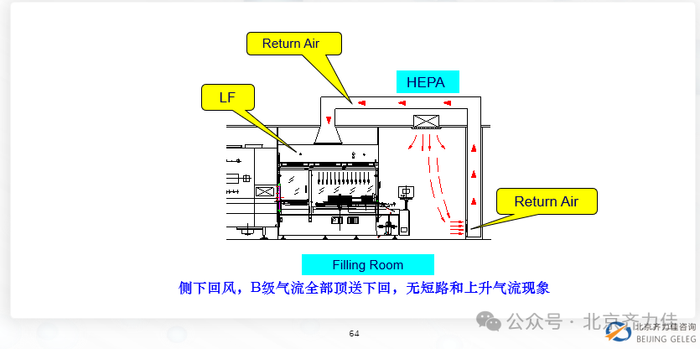
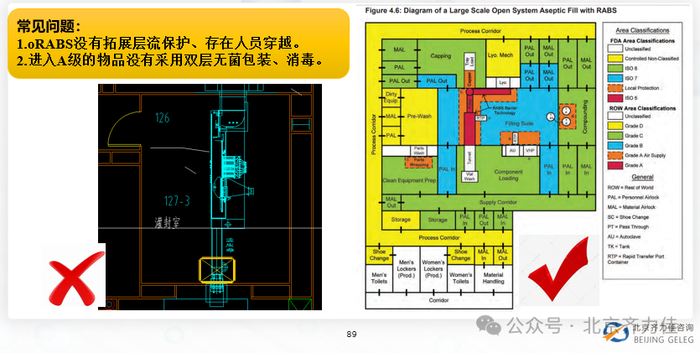


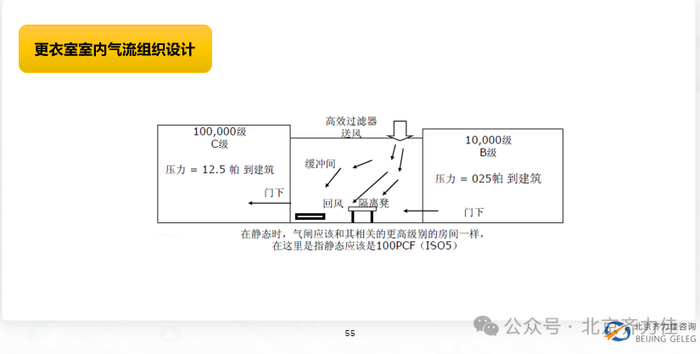

质量管理差距:
我们帮助客户进行全面的差距分析,找出上百条差距,依据风险大小逐步分析确定整改优先级。
2025版GMP无菌附录(征求意见稿)与之前的版本相比,有以下主要变化:
1. 内容更加丰富全面
章节条款数量增加:2025版GMP无菌附录(征求意见稿)共12个章节235条,约4万字,而之前的版本仅有81条、约1万字,内容大幅增加。
新增概念和要求:首次引入了污染控制策略(CCS)的概念,增加了药品质量体系(PQS)、质量风险管理(QRM)等原则性要求。同时,新增了细菌截留试验、限制进入屏障系统、首过空气、平衡时间、等动力学取样头、本地分离菌等术语定义。
2. 技术要求更加细化
生产技术要求更新:在生产特定技术章节中增加了成型—灌装—密封(FFS)技术、密闭系统、一次性使用系统等新技术的要求。例如,明确了灭菌后过滤器完整性检测(PUPSIT)的强制实施,推动企业从依赖终端检验转向过程控制。
洁净区动态监控要求提高:进一步细化了洁净区动态监测标准,如A级区悬浮粒子监测需覆盖灌装全过程,并允许在“产品本身产生粒子”时有限度豁免≥5.0μm粒子超标。
3. 质量管理理念深化
强调全面质量管理:更加强调对无菌药品生产质量进行全面管理,从整体上对风险进行梳理控制。要求企业建立污染控制策略,主动识别、科学评估、全面控制潜在质量风险。
鼓励采用新工艺新技术:鼓励企业采用一次性使用系统、密闭系统、机器人系统、快速检测技术等新工艺、新技术,以降低产品受到人员、环境等污染的潜在风险。
4. 人员管理要求提高
关键人员资质要求提升:对生产管理负责人和质量管理负责人的学历要求由现行的大专以上提高到本科以上,规定需要具备的相关管理经验。
人员操作规范更严格:对采用手工操作的情形提出更严格的标准要求,如在冻干机灭菌频率、无菌工艺模拟频次等方面均针对自动系统和手工操作设定了不同的标准。
5. 与国际标准接轨更紧密
参考国际标准制定:本次修订稿以PIC/S附录1为基础,并综合考虑我国现行附录要求及行业实践。FDA数据完整性指南形成协同。
适应国际监管趋势:近年来无菌药品生产相关技术发展迅速,国际上WHO、PIC/S均先后参考欧盟标准出台了相应无菌药品附录,三个版本实质内容基本一致。本次修订有助于我国顺利加入PIC/S。
北京齐力佳
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多