人工智能驱动的AlphaFold以及精度接近AlphaFold的RoseTTAFold的诞生令学术界大为震撼。人们对其所提供的可能性感到欣喜,不可否认其可能引起相关领域的重大进展。AlphaFold的强大的结构预测能力也被誉为有望改变药物的发现过程,这一强大的人工智能技术,在PBD数据库中学习实验结构并利用多序列比对,产生高度精确的预测。在计算结构生物学中,AlphaFold的模型可以作为仿真的输入,揭示激酶激活、自我抑制、致癌突变、蛋白质-蛋白质关联建模(对接)等机制。AlphaFold有助于解决晶体或冷冻电镜结构,并进行广泛的功能和结构特性统计分析。AlphaFold在深入了解正常生理条件和疾病下的蛋白质功能方面具有很大的优势。对于药物发现而言,情况更加复杂。最近的研究蛋白质的结构是多态性的。
蛋白质其实并非刚性的结构快照。混合的结果反映了它们的结构灵活性和构象异质性。最重要的是,药物结合口袋暴露在溶剂中。活性位点为了保持催化活性必须具有灵活性。几十年前,在比较嗜热和嗜温蛋白质时已经发现了这个独特的问题。进一步对活性位点进行详细研究表明,极性催化残基的曝光面积有限。它们被精确定位并固定在位,其具有较低的温度因子,并通过主干和侧链组之间的氢键进行结合。
尽管如此,酶家族成员之间有着多样性的选择性,这是通过少量残基的改变、静电作用和涉及非保守外环的动态波动来实现的。在激酶中,深部疏水性口袋中的ATP竞争性抑制剂通过利用ATP位点和周围口袋中的氨基酸之间的差异来实现其特异性。这种灵活性对于AlphaFold来说是具有挑战性的。AlphaFold学习了PDB中的结构,这些结构大多数都很好地折叠、紧密包装且稳定。而活性位点并不遵循蛋白质折叠的主要原则,使它们被人工智能驱动的方法来捕捉变得困难。
AlphaFold在药物发现中面临的一个巨大障碍是蛋白质存在于相互转化的构象系列,而晶体结构和AlphaFold的模型提供的是结构快照。近二十年前,。在晶体结构中,PDB文件列出了残基的B因子,这些因子是它们运动程度的指示。AlphaFold会给不同片段预测结构附加置信水平并对模型进行排名。虽然对用户有用,因为通常选择最高得分的解决方案,但这种排名不是基于物理学的,也不类似于动态构象状态。AlphaFold本质上无法扫描广阔的构象空间。这就好比违背了其本质特性。
AlphaFold在药物发现中能否有用?在接下来的讨论中,我们考虑了竞争性药物和变构药物两种类别。使用AlphaFold的模型可以为两种类型的药物提供合理的起点进行分子动力学模拟,提供更精细和准确的模型。在这里,我们考虑它们是否可以超越这一点,允许实际应用于虚拟对接筛选。竞争性药物和变构药物应当稳定并特异地锚定在其相应的蛋白质口袋内。然而,它们的作用机制完全不同。这表明它们与口袋内残基相互作用的某些性质是相似的,而其他方面则有所不同。在这两种情况下,其与相应结合口袋的特异性和亲和力应该很高,这就解释了搜索变构药物结合位点的方法相当成功的原因。
然而,处理变构位点检测的方法受到必须先识别出引起变构信号的药物-蛋白质相互作用的限制。迄今为止,没有一种方法能够做到这一点。此外,最近的研究显示,亲和力更高的位点不一定会导致功能上优越的药物结合。小分子整合素aIIbb3和a4b1的抑制剂稳定了高亲和力构象(图1),仍然导致部分激动和临床后期试验失败。有效变构药物发出的信号应当传播到活性位点,并改变涉及亚基(或辅因子)结合的残基的构象。
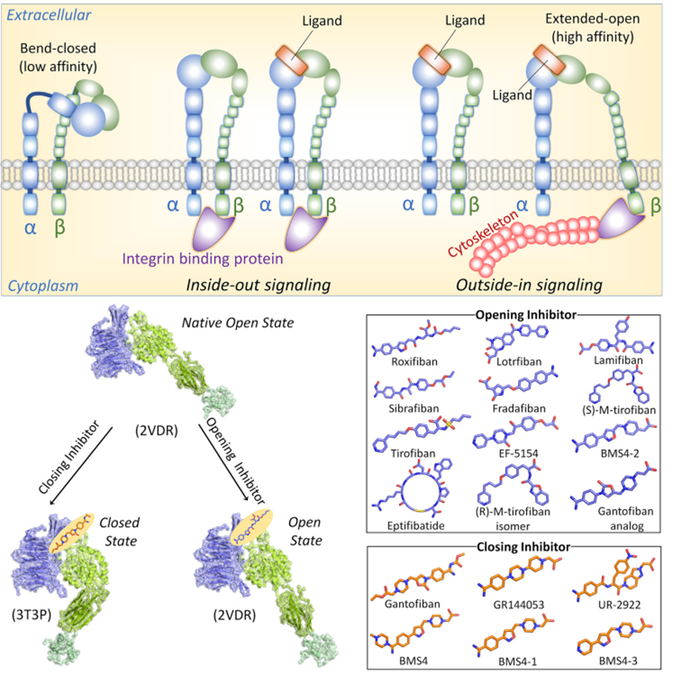
值得注意的是,在酶家族中,利用最高效的配体来发现活性降解化合物是一种无效的方法。在接下来,我们澄清正向和变构药物的不同机制,以及它们与其结合口袋中残基相互作用的共同和不同的描述符。然后,我们考虑如何根据它们的特性以不同的方式使用AlphaFold来发现这两种类型的药物,同时认识到蛋白质采用不同的结构,动态变化的群体。
目前绝大多数可用的药物都是正构药物。它们结合在活性位点上,并阻碍底物或辅因子的进入(图2a)。正向药物要发挥作用,其亲和力必须高于自然配体,这会带来一个问题,因为自然配体的亲和力可能非常高,如在Ras中的GTP的亲和力是皮摩尔级别,激酶中的ATP也具有非常高的亲和力。虽然可以通过更高的药物剂量来克服这个障碍,这会导致药物产生毒副作用,因为高亲和力的药物可以结合到结构相似的活性位点。相比之下,变构药物会结合并促进引起微小或明显构象变化的波动,从而影响活性位点。蛋白质由互相转化的构象系列组成。变构药物的结合改变了一些构象与其他构象的相对稳定性,导致蛋白结构种群的重新分布。这可能会阻碍自然配体的结合。正向药物和变构药物的不同机制预示着它们的可能结果。两种药物类型的不同作用模式也表明它们可以相互配合使用。变构药物可以改变药物耐药突变所形成的活性位点的构象,从而使被阻塞的正向药物得以结合(图2b)。
变构药物药物通常,但并非总是在保守位点结合。同时,这种缺乏保守性会在检测时带来问题。它应该是一个药物结合口袋,由化学上有利的残基组成。它还应该动态连接到活性位点, 并具有适当的信号传播路径将它们连接起来。结合于此处的药物必须特异地并稳定地锚定在那里,以便具有足够长的药物作用时间来发挥作用。
发现变构口袋具有挑战性,变构与构象和功能转换有关。在这种情况下,AlphaFold无法处理如此复杂的情况。执行特定功能要求蛋白质填充不同的构象状态,这可能无法被AlphaFold识别或建模,因为它可能在PDB中未被填充。在一篇1991年的里程碑性论文中,Frauenfelder、Sligar和Wolynes描述了蛋白质不仅存在于两个状态(活性和非活性状态)中,而是存在于各种状态的群体中,它们的种群与其相对能量水平有关。
自由能地形描述是强大的,因为它映射了蛋白质可以填充的所有可能构象。但是,地形描绘的图像是静止的。在我们的实验验证动态合奏地形描绘中,动态转移可以由各种事件引发,包括药物结合、正向和非定向作用。然而,正向药物引起的干扰是无关紧要的,因为药物已经阻塞了活性位点,而由非定向药物结合引起的干扰则是变构药物作用的核心。
AlphaFold对干扰是无动于衷的。在特异性和亲和力方面,两者对于两种药物类型都至关重要。有效药物应该具有有利接触的基团。但在两种情况下,高亲和力并不保证高特异性。正如下面讨论的那样,如果对接程序中的评分函数足够好,AlphaFold可以提供特异的高亲和力相互作用。
“好”的AlphaFold预测为基于物理的分子动力学模拟提供了合理的起点,从而生成了两种药物类型的高质量模型。问题在于AlphaFold是否能够超越这一点做出更大的贡献。AlphaFold的预测是静态模型,可能捕获不到关键残基的活性位点、侧链和骨架的构象。此外AlphaFold并不考虑活性和非活性状态的生物学和差异稳定性。这阻碍了药物发现,通常有利于活性状态,尽管像K-Ras4BG12C AMG510 (sotorasib)抑制剂和肿瘤相关酪氨酸激酶cAbl、c-Kit和血小板源性生长因子的突变体的Imatinib成功案例证明并不总是如此。AlphaFold的最高置信度的模型很可能处于PDB中的非活性状态。
这引发了一个问题,即采取什么策略来利用AlphaFold在药物发现提供更多的帮助。候选药物的体外对接可能不会选择非活性状态模型,因为它们相对稀疏和活性位点描述不准确。无序区域的频繁存在进一步混淆了这些工作。AlphaFold未来的准确性可能会得到改善,并且可能会获得预测与配体复合物的额外功能。然而,考虑到小分子-蛋白质数据集对于深度学习可靠性的影响以及涉及不同原子类型的大量小分子交互,在即将出现的额外复杂因素中,仍可能遇到障碍。提高更高准确性的额外数据以及新的机器学习模型可用于处理化学,并注入真实世界的物理有效性。化学和生物物理学,即原子类型和相互作用、有利拓扑、侧链和相互作用能量是对接策略的关键属性。
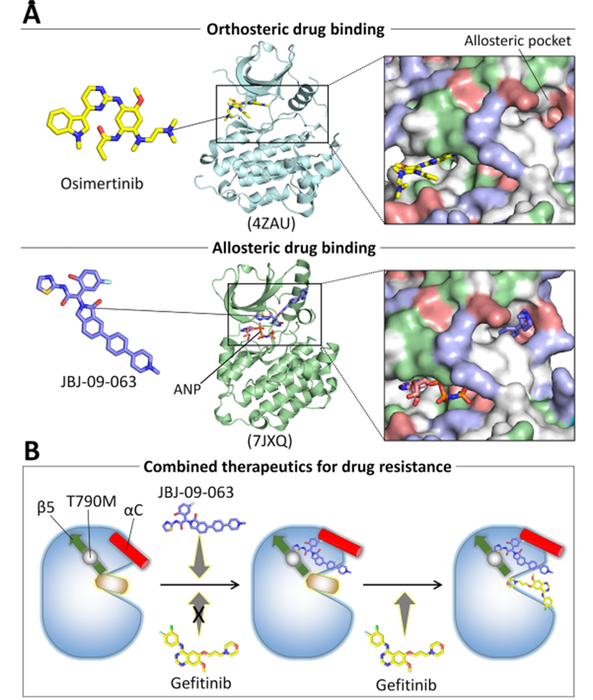
图2. 正构抑制剂和变构抑制剂作用机制
蛋白质由动态集合体组成。AlphaFold本质上是刚性的。在药物发现中处理灵活性的一种简单方法是改变对碰撞中何为“匹配”的定义,允许更大的阈值。这可能适用于侧链和轻微骨架,但不适用于更大的构象变化。或者,可以将分子动力学模拟应用于AlphaFold模型,并使用所得到的聚类构象作为对接方法的输入。模拟可能会或可能不会采样所有相关状态。这取决于分子大小、构象转变的壁垒、构象变化的程度、相对稳定性和模拟时间尺度。
AlphaFold与药物进行虚拟对接模拟时观察到的高误报率可能部分原因是该模型被严重偏向于PDB中非活性状态。通常,非活性状态比活性状态更稳定(图3a)。这是细胞的要求。具有有利活性状态的激酶,往往是由于突变导致其稳定并/或使非活性状态不稳定,可导致癌症。一个可能的方法是改变AlphaFold的非活性构象偏见。对于激酶则涉及DFG-in/out构型的转变(图3b)。非活性状态和活性状态之间的差异很常见,并且最近已经获得了这些状态的选择性结构模板用于5-HT5A受体的建模和对接。
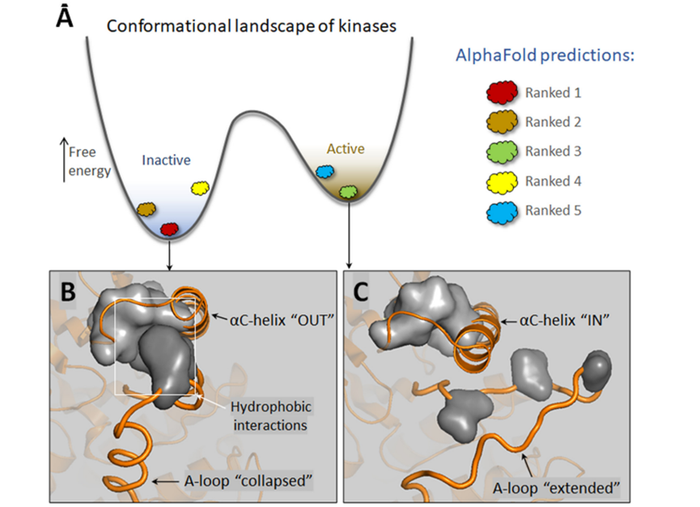
因此,在不进行构象空间详尽枚举的情况下,考虑蛋白质状态和灵活配体是前进的可能性之一。机器人引导的灵活模式检测过程是另一种方法。确定性方法可以在模式检测过程中考虑灵活性,这可以利用生物学知识,例如蛋白激活/非活动状态示例。酶具有单个活性状态和多个非活性状态。过滤和平衡AlphaFold的模型可以改善对接中真阳性与假阳性的比率。基于更广泛的生物学的确定性策略,可以构建蛋白质和药物构象空间的模型,提供了一种现实可行的方法。确定性状态也适用于变构药物的发现。
不仅AlphaFold模型是刚性的,X射线结构也是刚性的。X射线晶体学中的灵活性通过B因子和未分配残基/原子坐标来表示。然而,X射线晶体学仍然被认为是药物发现中最重要的工具之一。这种明显的矛盾可以通过回顾蛋白质可以与配体同时结晶,捕获构象变化来缓解。单晶结构不能捕捉变构转变。灵活性通过构象波动来表征。它涉及多个构象,可以通过克服动力学壁垒相互转化。到目前为止,唯一基于相同输入生成集合的方法是分子动力学模拟。
模拟可以以非常精细的时间分辨率捕获蛋白质的行为,使原子处于不断运动状态。已有研究讨论了利用机器学习指导模拟生成构象数据,以增强和扩展AlphaFold的应用性。进化尺度建模或ESMfold也预测刚性模型。该预测依赖于采用自我关注机制的新深度学习语言模型。与AlphaFold不同,它不依赖于多序列比对。仅查看单个蛋白质序列可以提高其速度,同时保持高质量的预测。但是,如果使用,准确性可能仍然会进一步提高。
即使是刚性的,几个研究小组已经尝试通过为AlphaFold提供浅层多序列比对或不同输入结构的学习集来迫使其预测多个状态。预测的模型在原子分辨率上捕获了GPCR激活后的主要结构变化。在将AlphaFold与实验X射线和溶液中测量的剩余偶极耦合进行严格对比时,在三组输入下生成了三组AlphaFold模型。第一个是通过利用标准的AlphaFold输入结构数据库生成的。第二个AlphaFold运行的输入排除了2020年1月1日之后沉积的X射线结构,第三个排除了与SARS-CoV-2的冠状病毒同源二聚体3C样蛋白酶,Mpro同源的所有结构。这些最新的发展表明,预测刚性模型的AlphaFold可以通过多种方式产生结构变异体。然而,目前还不清楚是否通过提供AlphaFold来预测多个状态,例如,一个浅的多序列比对,预测药物发现所需要的生物相关活性激酶状态。
现在怎么办?药物发现是一个紧迫的问题,强大的预测工具指日可待。同时,我们需要认识到AlphaFold能做什么,不能做什么。AlphaFold对刚性蛋白质结构进行建模,同时通过可靠性分数来考虑柔性。它利用序列和结构相似性将实验结构中的小分子和离子"移植"到预测的蛋白质模型中。增强活性状态候选群体可以有效地用于正构和变构药物的发现。机器学习算法的进步可以高精度地捕获蛋白质的活性非活性动力学。在最近的一个发展中,使用注释的活性/非活性GPCR构象作为模板,启用了多状态预测协议,AlphaFold2以非常高的精度预测活性或非活性状态。值得注意的是,预测模型在原子水平上准确地捕获了GPCR激活时的主要结构变化,表明通过适当的学习模型,活性/非活性预测是可行的。
有人问道:“在这个计算能力和成就无与伦比的时代,如AlphaFold预测每个蛋白质的结构,化学家将在什么时候看到药物发现方式的根本改变? ”,虽然人工智能很难在短时间内战胜人类多年积累的经验,以及对结构和细胞复杂性的洞察力。然而,快速增加的数据加上算法的进步,并以人类的经验和直觉为指导,相信这并非无可能的事情。
Nussinov R, Zhang M, Liu Y, et al. AlphaFold, allosteric, and orthosteric drug discovery: Ways forward[J]. Drug Discovery Today, 2023: 103551.
内容来源:智药邦
责任编辑:胡静 审核人:何发













评论
加载更多