辅料的关键质量属性研究与控制
在制剂制备过程中,药用辅料选用是否得当会影响制剂的安全性、有效性、稳定性、经济性和顺应性。
本文通过围绕辅料的关键质量属性,通过几个案例展示了辅料功能和性质对制剂研发的影响,并总结了辅料质量属性的分析技术,旨在提示广大研发人员重视辅料的质量研究与控制。
1、什么是辅料的关键质量属性(CQA)?
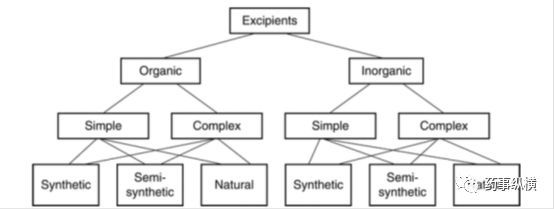
辅料可以有多种分类方式,如下图可见,包括有机辅料、无机辅料,每类别根据结构不同都可以分为简单(结构简单、明确)和复杂(结构复杂、不明确)的,根据来源不同,分为合成、半合成、天然的辅料。
关键质量属性是物理的、化学的、生物的、微生物的属性和特性,这个属性应被限定在一个适当的限度、范围内,以确保产品的质量。
对于原料药而言,可能包括质量标准范围内的,如有关物质、含量、异构体、水分、干燥失重、残留溶剂、溶解性等,还有可能包括与制剂的溶出度、生物利用度、稳定性、工艺等相关的晶型、粒度、晶癖。
而对于辅料而言,一般包括杂质、粒度、粘度、pH、分子量分布等特性。与原料不同,辅料的关键质量属性有时很难发现,由于受到API(性质、剂量大小)、处方(组成、比例)、生产工艺等的影响,某种属性(例如粘度)对于某药物可能是关键的,而对另一种药物,或另一个不同剂量的药物而言,可能就不是关键的,也就是说辅料属性的发现和确定是否关键与很多因素有关,如果在研发初期研究不充分、没有发现、没有总结,可能就不会发现辅料的关键质量属性,即使一次通过生物等效性试验(BE),也难保后期放大、持续生产、临床安全性、有效性方面不出问题。
因此,在研究过程中,我们还应该把更多的精力放在系统研究辅料特性上,细致思考和观察,把辅料的CQAs与成品的处方组成、生产工艺和CQAs结合起来,找到其中的因果关系。
2、辅料性质差异从何而来?
不难理解,与API一样,辅料的理化性质、机械性质的差异也是由其生产工艺决定的,不同的生产工艺(或参数)会生产出不同性质的辅料。如果辅料生产的起始物料、生产工艺(如全合成、半合成、发酵)、生产设备、生产规模、工艺参数等发生了变化,那么辅料中也就可能产生新的工艺杂质,也可能导致某些理化性质发生变化,甚至引起机械性质、粉体学性质的变化,例如不同pH、不同粒度等。
一般来讲,对于通过发酵、降解工艺生产的辅料,其发生质量差异的可能性比全合成的辅料要大。因为前两者的生产工艺有时较复杂,工艺参数控制往往较宽,而后者如果起始物料不变,工艺参数调整的空间较小。
在原辅料相容性过程中,在不同厂家辅料的筛选中,在稳定性的放样考察中,要特别注意研究和总结不同性质的辅料对有关物质、含量、溶出等CQA的影响,然后去分析辅料的哪些属性会引起这种结果,是水分、pH、杂质、过氧化物、粒度还是其他。对于特殊工艺制备的制剂(如缓释制剂),特殊BCS分类的药物(如BCS II或IV类)来讲,辅料的特性研究更加关键。研发中,要关注不同供应商来源的同种辅料、相同厂家的不同批次辅料之间的差异,尽可能多了解辅料的生产工艺。
辅料CQA能否发现,与人员水平有关、工艺有关、还与所选择检测方法和技术有关。
3、影响制剂CQA的辅料特性
辅料对制剂成品质量属性、生产工艺的影响是通过辅料的功能、辅料的质量以及辅料与辅料、辅料与药物的相互作用来实现的。辅料的性质可分为物理性质和化学性质两大类。在物理性质中,常见的性质例如粉体学性质(流动性、可压性、粒度分布、形态等);化学性质中,常见的性质例如化学结构式、取代度、杂质等。目前有非常多的关于辅料特性差异的报道,作者仅仅列出几个案例来说明问题,其他代表性案例,读者可自己可通过文献查找,不再一一列举。
案例1:不同粉体学性质辅料对直压工艺产品质量的影响
Parteck®M200、M100 直压甘露醇和直压甘露醇C、D 这四种不同型号的甘露醇与主药相容性好,但不同型号的甘露醇之间还是存在一些差异[1]。
表1. 粉体学测定结果
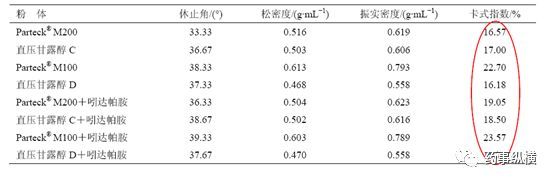
一般来说,卡式指数在25%以下时流动性较好,卡式指数越小,粉体的填充性越好,相反卡氏指数较大,可压性好。通过表1可以看出除Parteck® M100 直压甘露醇外的3 种型号甘露醇及其混合药物后测得的卡式指数均小于20%,而Parteck® M100 直压甘露醇及其混粉的卡式指数也不大于25%,在填充性和可压性方面各有所长。
然而,在不同溶出介质中的溶出曲线表明,在水中,含Parteck® M200、M100直压甘露醇的模型药物吲达帕胺的溶出结果优于直压甘露醇C、D,而在其他3 种溶出介质中的4条溶出曲线趋势一致,说明药物与Parteck® M200、M100 直压甘露醇混合压片后的溶出度具有一定优势。
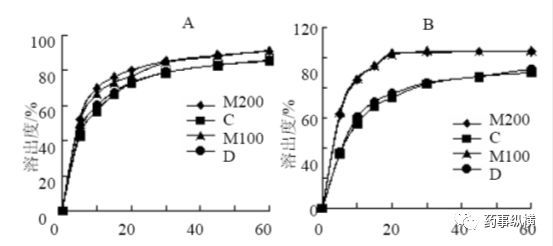
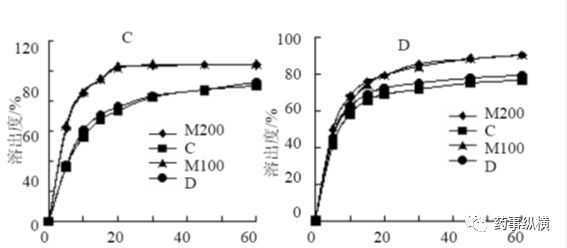
图1. 片剂在水(A)、0.1mol/L 盐酸(B)、pH 4.5 醋酸缓冲液(C)和pH 6.8 磷酸缓冲液(D)中的溶出曲线
通过这个案例可知,相同种类辅料不同型号、不同粉体学性质的辅料在特定处方、工艺条件下,对制剂成品的关键质量属性影响不同,因此对于粉末直压工艺而言辅料的粉体学指标是一个关键因素。
案例2:不同粒度甘露醇对成品含量的影响
采用粒径较大的物料甘露醇200SD和氯雷他定混合粉压片,发现压片过程中的硬度、厚度、重量差异等均符合要求,但压片结束时片子的含量偏高,是什么原因呢?
原来200SD在混合粉中先从料斗中流出,而粒径较小的主药粘在料斗壁上,最后流出,导致压片结束时饲料器及片子含量偏高。当换用粒度较小的甘露醇100SD 时,由于100SD的粒径和主药差异不大,因此并没有出现分层的现象,示意图如图2 所示[2]。
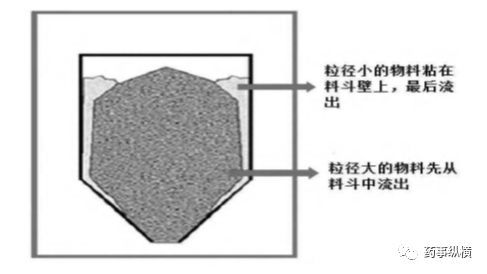
图2. 粉体在压片过程中料斗内的流变学
作者在对压片过程中模孔和片子的数据进行分析时发现,甘露醇100SD和200SD混合粉体表现出了相同的现象:模孔内的含量均比同时间的片子含量偏高。产生这种现象的原因是什么呢?作者认为,对于直接压片工艺,粉末并未经过制粒过程,粉末分子间仅有微弱的范德华力连接,很容易受到外界的影响,上冲在压模孔里的物料时,造成了粒径较小的主药飞散出模孔而使最终片子的含量相对偏低。
通过这个案例可知,相同种类不同粒度的辅料,在特定药物、处方、工艺条件下,对成品的生产工艺和关键质量属性的影响不同,因此“粒度”是一个关键因素,在一个选定的制剂工艺条件下,需要对辅料提出“特殊要求”。
案例3:不同来源SLS对制剂工艺的影响
在设计难溶性药物口服固体制剂时,制剂人员面临着极大的挑战,特别是高剂量药物。提高溶出度和/或溶出速率是提高难溶解性药物口服生物利用度的必要条件。目前有许多方法可以提高溶出,如采用酸化剂或碱化剂调节pH值、络合物、固体分散体、制备成无定型药物、减小粒度以及使用表面活性剂等。表面活性剂可以在提高药物溶解度方便发挥一种或多种作用,如改善药物的润湿性和溶解性,并减少或防止药物沉淀。从生产角度而言,使用表面活性剂提高溶解度比其他非常规剂型(如固体分散体)更适合生产与降低成本。
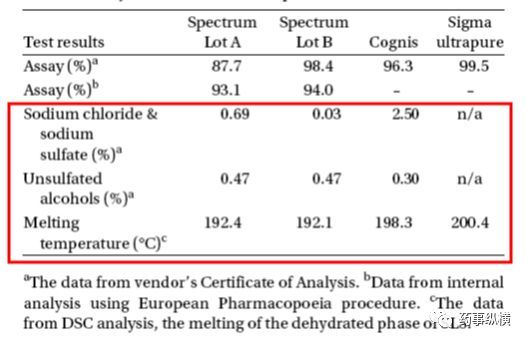
十二烷基硫酸钠(SLS)是一种很常用的阴离子型表面活性剂,具有很强的增溶能力,用在固体口服制剂中提高难溶性药物的溶解度。除了上述作用之外,SLS还会影响口服固体制剂的生产过程。不同来源的SLS含有不同含量的表面活性杂质,其中大部分是同源的醇类,其表面活性在SLS的两个数量级以上(见表2)。这些已知或未知的杂质可能有助于表面活性剂的功能,并改变SLS在溶出介质或固体口服制剂中提高药物溶解性时的增溶作用。不同来源的SLS可能具有不同的物理性质,如熔点、粒度、微观形态,这可能影响药物的溶出和生产过程[3]。
表2. 不同来源的SLS检测结果
不同来源的SLS的扫描电镜图如下所示,Spectrum公司SLS大多数颗粒具有更少的球形形状,长度大于25μm,一些聚集体大于150μm。Cognis公司的SLS大多数颗粒是较大、多孔、不规则的聚集体。Sigma公司的SLS颗粒呈薄片状,表面有层状结构,颗粒的直径约为2μm至数百微米。三个公司的SLS粒度分布测定结果差异很大。

图3. Spectrum公司的SLS扫描电镜图

图4. Cognis公司的SLS扫描电镜图

图5. Sigma公司的超纯SLS扫描电镜图
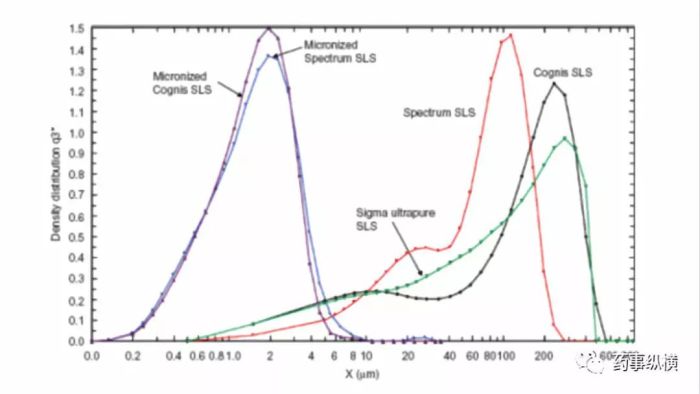
图6. Spectrum、Cognis、Sigma公司的SLS粒度分布图(未微粉、微粉)
在湿法制粒中,不同来源和不同粒度的SLS所需的水量和制粒时间也不同。含有SpectrumSLS的辅料制粒时的用水量比其他两个来源的SLS少。因此在固定制粒时间时制粒所需的水量与SLS的表面张力的顺序相同:Spectrum < Cognis < Sigma(超纯)。
表3. 不同来源和不同粒度的SLS所需的水量和制粒时间
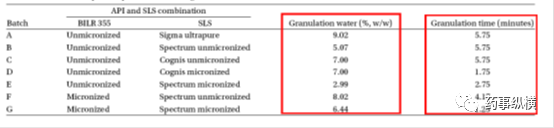
根据上述案例可见,在进行含有SLS的制剂处方工艺研究中有必要研究不同来源SLS的性质、粒度分布等性质,必要时制定辅料的内控标准。
案例4:辅料化学结构式与功能的相关性
某药物的片芯辅料之一为all-rac-α-Tocopherol,中文名为α-生育酚,结构式中含有酚羟基,作为抗氧剂使用。但有时研发人员误将其认为是维生素E而使用了中国药典中的原料药维生素E作为抗氧剂使用,从结构式中可见由于药典中的维生素E为α-生育酚的醋酸酯,因此已没有抗氧化作用了。所以,使用辅料时应注意辅料的化学结构和其作用的关系。
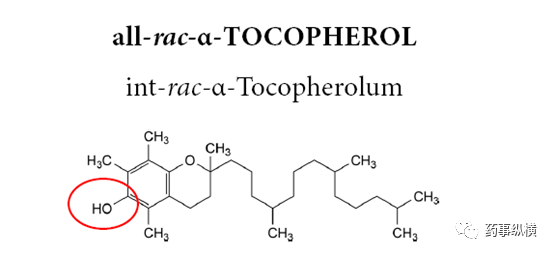
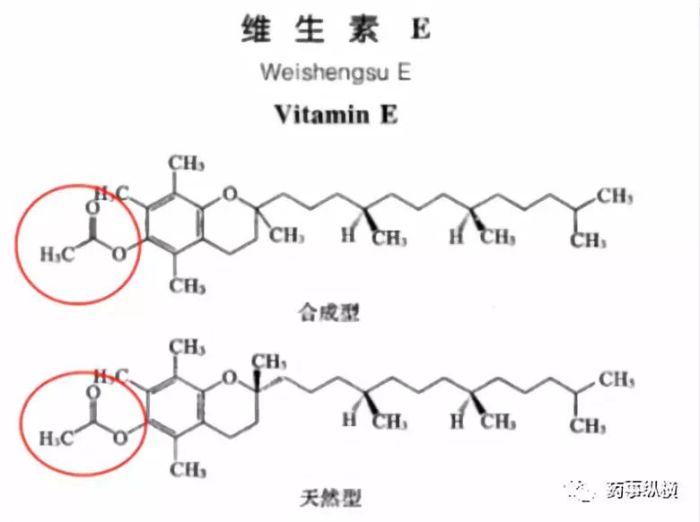
案例5:辅料中的活性杂质
辅料中也有很多类活性杂质,这类杂质可以与API发生反应。以过氧化物为例,聚山梨酯、聚乙烯吡咯烷酮(PVP)、聚乙二醇(PEG)、羟丙基纤维素(HPC)等是含有过氧化物的辅料,如果根据文献报道或研究发现API容易被氧化,由于不同的API对氧化的敏感性可能不同,那么在处方筛选阶段就需要设计相应的实验来研究论证这些辅料中不同过氧化物含量对制剂中杂质的影响,根据数据结果来确定该辅料中过氧化物需要控制在什么限度才能保证制剂的质量和稳定性,保证杂质在整个货架期内的增加趋势不高于原研制剂。
辅料中的过氧化物包括过氧化氢(H2O2),氢过氧化物(ROOH),有机过氧化物(ROOR’)等。过氧化物常用于聚合物的生产,在纯化过程中很难被完全去除,此外,在聚合物的降解时也可能会产生过氧化物。不同的生产条件、纯化工艺、不同存放时间、不同生产厂家、以及相同厂家不同批次之间,过氧化物的含量都可能存在较大差异。
过氧化物可以通过以下三种途径与API发生氧化反应:亲核加成、亲电取代、金属存在下的自由基反应。如果发现API中的氧化杂质增加,且处方中包括上述辅料应进行有针对性的研究和控制。
醛类、有机酸(如甲酸、乙酸等)、还原糖、元素杂质等辅料中活性杂质的控制综见美国基因泰克公司的综述[4]。
4、辅料CQA的分析技术
从上述案例中可见,辅料变异有时会显著影响自身的性质和功能,也会对制剂的关键质量属性和工艺的选择重要有影响。我国仿制药一致性评价申报资料中的P.4部分就要求对辅料的控制进行研究和说明。因此,在仿制药研发中,不但制剂人员应该对辅料有深入了解,分析人员也应该掌握评价辅料特性的分析方法,以促进对QbD药品研发先进理念的理解。
从上文分析可知,辅料差异的来源包括:source to source,site to site,batch to batch, supplier to supplier,year to year等。为了表达这些差异,我们可以从“微观”和“宏观”两个角度进行分析。
辅料的微观属性分析技术。X射线粉末衍射法、DCS法、TGA法、NIR法、固态核磁SSNMR、KFT或干燥失重法、pH测定法等方法常用来对辅料的微观属性进行分析,如结构式、晶型或无定型、熔点、水分、酸碱度等。其中,SSNMR法区分辅料化学结构、NIR法在区分辅料批间差异方面具有重要应用。
辅料的宏观属性分析技术。辅料粉体学特性的测定方法,如流动性、堆密度、压缩指数、粒度分布、扫描电镜(SEM)等指标都有相关的检测技术,详见USP<1174> POWDER FLOW粉体流动性等其他检测技术章节。
当辅料的来源、生产工艺、稳定性等发生变化时,采用上述分析技术进行综合分析,往往可以发现辅料性质的差异,当然辅料的检测方法也是需要进行方法学验证的。
5、辅料质控相关的指导原则和质量标准
ICHM7和ICHQ3D大家经常会用于成品的质量控制,如原料药或制剂。但是,对辅料的质量控制而言,也有特殊规定。例如ICHM7规定对已上市药品中使用的辅料、香精、着色剂等可不进行基因毒性杂质的有关研究,然而对于新辅料而言,ICHM7要求对首次在制剂中使用到的辅料进行安全风险评估,因此这类辅料需要关注遗传毒性杂质的控制。ICHQ3D用于原料药和制剂的金属杂质控制,对于辅料而言,也可能引入金属杂质,如Pt、Pd等,ICHQ3D同样适用于辅料的金属杂质研究,常用的方法为ICP-MS。
此外,EP、USP-NF、JP等国外药典标准也经常收载ChP没有收载的辅料标准,在对辅料标准进行分析研究时,应考察将其汇总对比,详细标准检测方法和限度,根据对制剂产品关键质量属性的影响,制定相应的控制指标和控制方法、限度标准,从物料的源头上来确保产品质量的稳定性和均一性。
6、QbD研发理念与辅料控制
QbD理念通过ICHQ8、Q9、Q10指导原则进行了表达,QbD强调对制剂处方、工艺的理解和控制,其中重点概念包括目标产品质量概括QTPP、关键质量属性CQA、关键物料属性CMA、设计空间DS等。因此应该对原料、辅料、包材、工艺、设备、分析方法等进行充分的研究与理解,对今后可能出现变异进行提前判断。
对于辅料的选择,是药品研发中一个很重要的环节,辅料的选择影响生物利用度(BA)或生物等效性(BE)、药物稳定性以及生产重复性。有时,辅料选择时往往需要综合考虑很多问题,如为了提高生物利用度、生物等效性而牺牲一定程度的药物稳定性,这时就需要选择合理的包材或制定合理的控制策略来实现和保证。
把辅料的CQAs与制剂成品的QTPP联系起来,只要制定了辅料控制策略就能够保证产品符合要求,而为了完成这种“过程中的控制策略”,首先需要结合国内外药典标准对辅料的基本检测指标进行分析;其次需要结合辅料的厂家报告和自检结果分析存在的差异;第三需要结合制剂处方工艺研究寻找辅料的CQAs并制定适用于特定药物的内控标准。
辅料虽然无药理活性,但却是制剂的重要组成部分,缺少辅料制剂的性能是无法实现的。药品中使用到的辅料的性质往往是发生变化的,这些变化往往来源于生产工艺的变化。辅料的某种特性的变化会影响到制剂的性能和功能,应充分理解了这些变化对制剂关键质量属性的影响。多种辅料的分析检测技术为评估这些变化提供了方法,应注意积累和总结。此外,辅料的包装、贮藏条件对辅料质量的影响,例如光、氧、湿、热等因素都会促进辅料中过氧化物的形成,聚维酮等辅料使用中需注意包装的合理性和完整性。
可以预见,随着我国仿制药一致性评价工作的深入开展,人们对辅料关键质量属性的研究和控制将会得到前所未有的重视。
参考文献
[1] 白璐,王博,吴疆,任晓文.不同型号甘露醇对吲达帕胺直接压片的影响研究[J].现代药物与临床,2014,29(4):338-341
[2] 赵存婕.探讨不同粒度的甘露醇对于直接压片产品的影响[J].黑龙江医药,2014,27(3):559-562
[3] Dongmei Qiang, Jocelyn A.Gunn, Leon Schultz & Z. Jane Li.Evaluation of the impact of sodium lauryl sulfate source variability on solid oral dosage form development [J]. Drug Development and Industrial Pharmacy, 2010; 36(12): 1486–1496.
[4] Kelly Zhang, Jackson D.Pellett, Ajit S. Narang, Y. John Wang, Yonghua Taylor Zhang. Reactive impurities in large and small molecule pharmaceutical excipients-A review [J]. Trends in Analytical Chemistry,101 (2018) 34-42.
文章来源:药事纵横
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多