化学药 | 一文告诉你,新药是怎么从研发到上市的,收藏!
医药公司为什么要研发新药呢?
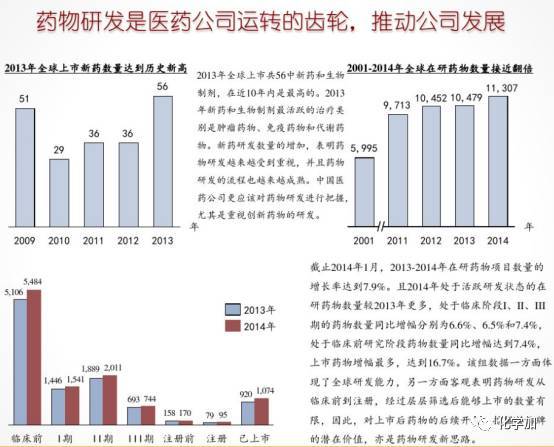
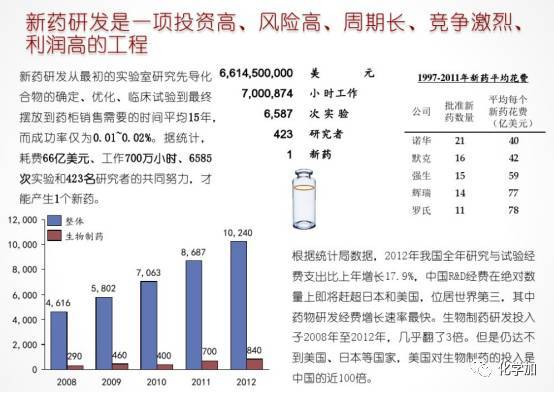
通过上图我们对新药研发的重大意义和复杂性有了一些了解,下面我们来具体说说新药研发的一些细节。
总的来说新药的研发分为两个阶段:研究和开发。这两个阶段是相继发生有互相联系的。区分两个阶段的标志是候选药物的确定,即在确定候选药物之前为研究阶段,确定之后的工作为开发阶段。所谓候选药物是指拟进行系统的临床前试验并进入临床研究的活性化合物。
一, 新药研究阶段
研究阶段包括四个重要环节,即靶标的确定,模型的建立,先导化合物的发现,先导化合物的优化。
1, 靶标的确定
在基础研究中,科学家们努力寻找特定疾病中发生作用的细胞和基因,以及针对特定生物参数和功能的化学或生物物质,希望能够发现其具有类似药物的作用。保守估计目前所有的药物治疗大概只覆盖了约700个药物靶标,在未来最少还有近10倍左右的药物靶标未被发现。药物的靶标包括酶、受体、离子通道等。
目前,较为新兴的确认靶标的技术主要有两个。
一是利用基因重组技术建立转基因动物模型或进行基因敲除以验证与特定代谢途径相关或表型的靶标。这种技术的缺陷在于,不能完全消除由敲除所带来的其他效应(例如因代偿机制的启动而导致的表型的改变等)。
二是利用反义寡核苷酸技术通过抑制特定的信使 RNA 对蛋白质的翻译来确认新的靶标。例如嵌入小核核糖核酸(snRNA)控制基因的表达,对确证靶标有重要作用。说的通俗一些,药物靶标的发现是基础科学的范畴,有必然性,但也许偶然性会更大一些。但是新药研发靶标的确定(因为我国研究一般都是跟在外国后面),需要做的就是在已知的靶标中挑选合适的靶标:这个阶段也称为靶标发现和靶标选择。靶标可以是单个基因、蛋白质或与许多不同疾病相关的蛋白质相互作用的通路。科学家通过各种方法来分离和识别特定的靶标,从而更好地理解靶标的功能以及与疾病之间的关系。然后设计或分离出与靶标相互作用的化合物。
2,模型的建立
靶标选定以后,要建立生物学模型,以筛选和评价化合物的活性。通常要制订出筛选标准,如果化合物符合这些标准,则研究项目继续进行;若未能满足标准,则应尽早结束研究。一般试验模型标准大致上有:化合物体外实验的活性强度;动物模型是否能反映人体相应的疾病状态;药物的剂量(浓度)——效应关系,等等。可定量重复的体外模型是评价化合物活性的前提。近几年来,为了规避药物开发的后期风险,一般同时进行药物的药代动力模型评价(ADME评价)、药物稳定性试验等。当然,到底是先建立模型还是先导化合物先产生,有点类似于先有鸡还是先有蛋的争论,在出PCC之前这是一个循环的过程。

3,先导化合物的发现
新药研制的第三步是先导化合物的发现。所谓先导化合物(leading compound),也称新化学实体(new chemical entity,NCE),是指通过各种途径和方法得到的具有某种生物活性或药理活性的化合物。因为目前的知识还不足以渊博到以足够的受体机制指导药物设计以使药物的合成不必使用预先已知的模型,所以,先导化合物的发现,一方面有赖于以上两步所确定的受体和模型,另一方面也成为了整个药物研发的关键步骤。一般来说,先导化合物主要有如下几个来源:对天然活性物质的挖掘、现有药物不良作用的改进以及药物合成心中间体的筛选等。目前,主要有两个获得新的先导化合物的途径。一是广泛筛选,这种毫无依据的方法在实际操作上其实是比较有效的。另外,先导化合物的合理设计近年来也越来越成为这一领域的热点。所谓合理设计,是指根据已知的受体(或受体未知但有一系列配体的构效关系数据)进行有针对性的先导化合物设计,这种方法有别于一般普遍筛选的显著特点在于目的性强,有利于各种构效理论的进一步发展,因此前途十分广阔。
说起先导化合物的广泛筛选,就离不开高通量筛选。
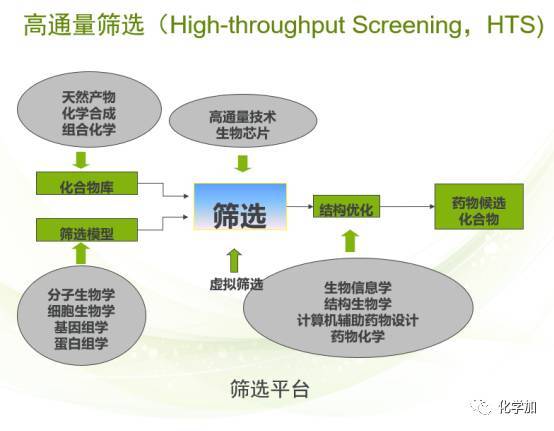
高通量筛选指以分子水平和细胞水平的实验方法为基础,以微板形式作为实验工具载体,以自动化操作系统执行试验过程,以灵敏快速的检测仪器采集实验结果数据,以计算机分析处理实验数据,在同一时间检测数以千万的样品,并以得到的相应数据库支持运转的技术体系,它具有微量、快速、灵敏和准确等特点。简言之就是可以通过一次实验获得大量的信息,并从中找到有价值的信息。
新分子一边合成出来,一边做各种生物活性,药代动力学,毒理这类的研究,找到我们需要的分子。
药动学参数:

毒性研究包括两类:急性毒性和长期毒性
急性毒性是指机体(动物)一次性或短时间内(24小时)多次接触外源性化合物后短期内所产生的毒性效应。
长期毒性是指机体长期连续或反复接触外源性化合物后所产生的毒性效应。
长期毒性一般包括一周或两周,更长时间包括4周,8周甚至数年。早期安全性评价只做一周或两周的动物试验。
长毒试验通过剂量爬坡获得动物对该药物的最大耐受剂量(maximum tolerated dose,MTD)。
毒性试验主要观察给药后动物的体重、摄食摄水量、症状、死亡、血液生化、肝活性、尿液以及动物出现死亡时进行组织病理切片。
药物化学家根据早期安全性评价的结果,确定是否进一步优化先导化合物
4,先导化合物的优化
在该阶段,生物学家和药物化学家一起努力使先导化合物更安全更有效。这是基于各步骤知识复杂和反复的过程。通常一个或多个先导化合物进行安全评价,合成和筛选。这就是所谓的类似物。类似物测试,其结果是与化合物结构变化相关的生物活性和药理数据。这些用于建立结构 - 活性关系(SAR)。新类似物将反馈到系统的下一个优化步骤中。最终得到优化的先导化合物,进入到临床前研究。进入临床前研究的化合物称为临床前候选化合物。
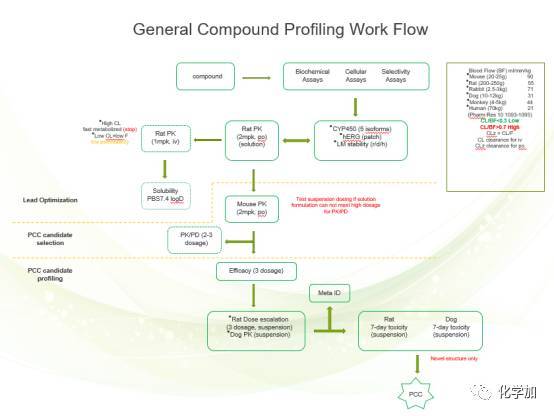
概括起来如下图所示:
二, 新药开发阶段
新药开发阶段如下:临床前试验,研发中新药申请,临床研究,新药申请。
临床前试验:由制药公司进行的实验室和动物研究,以观察化合物针对目标疾病的生物活性,同时对化合物进行安全性评估。这些试验大概需要3.5年的时间。研发中新药申请(Investigational New Application,IND):在临床前试验完成后,公司要向FDA提请一份IND,之后才能开始进行药物的人体试验。如果30天内FDA 没有发出不予批准的申明,此IND 即为有效。提出的IND需包括以下内容:先期的试验结果,后续研究的方式、地点以及研究对象;化合物的化学结构;在体内的作用机制;动物研究中发现的任何毒副作用以及化合物的生产工艺。另外,IND必须得到制度审核部门(the Institutional Review Board)的审核和批准。同时,后续的临床研究需至少每年向FDA提交一份进展报告并得到准许。然后就是临床一期,临床二期,临床三期以及上市后的安全性监督研究。
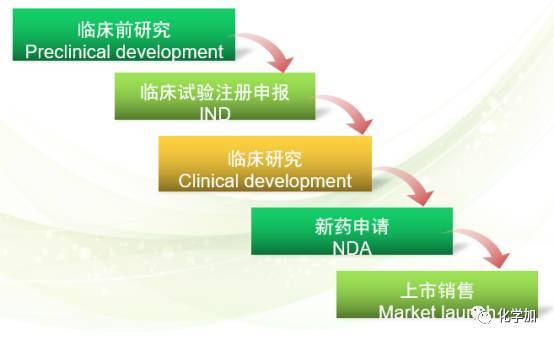
1, 临床前试验
临床前研究的主要内容:合成、生产和控制(Chemical、Manufacture & Control),制剂(Pharmaceutics),药理学(Pharmacology),药效动力学(Pharmacodynamics),药代动力学(Pharmacokinetics),毒理学(Toxicology),急性毒性试验(acute toxicity testing),重复给药毒性试验(repeat dose toxicity testing),长期毒性试验(long term toxicity testing),致癌性试验 (carcinogenicity toxicity testing),生殖毒性和致畸性(reproductive toxicity),基因毒性试验/致诱变性(genotoxicity/mutagenicity),毒代动力学(toxicokinetics)等。这里面的每一项内容都需要大量的时间和金钱的投入。
2, 研发中新药申请

3, 临床研究
临床研究一般包括四个阶段,即,首次试用于人类(健康受试者)的一期临床研究,试用于少数患者的二期临床研究,有上千患者参加的三期扩大临床研究,以及上市后进行实用验证的四期临床研究前三期为新药上市前必经阶段,第四期为药品上市后的监督性研究。
新药临床试验的分期及基本要求:
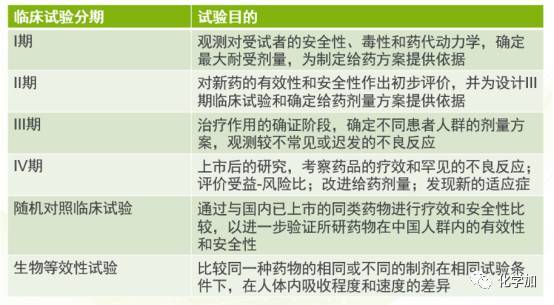
l期临床试验,一般为20~30例,有时会到100例。
II期临床试验,多中心临床试验,一般试验组不少于100例。
III期临床试验,更大规模的多中心临床试验,要求试验组不少于300例。如为双盲试验设计,则至少为300例;如为单盲试验设计(试验组和对照组并非1:1的比例),则只需满足试验组不少于300例。
IV期临床试验,不少于2000例。随机对照临床试验,多中心临床试验,一般不少于100对。如为多个适应症,则每个主要适应症的病例数不得少于60对。生物等效性试验(BE试验),一般为18~24例。根据药物的特性,可适当调整样本量的大小(变异越大的药物所需的病例数越多,目前国外最多有做到100多例的BE试验)。
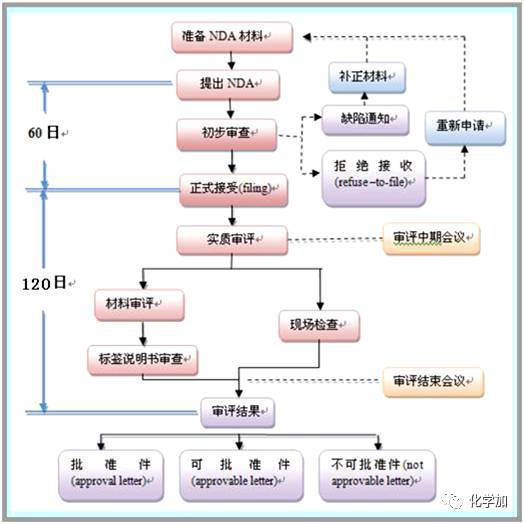
4, 新药申请
通过临床试验,公司将分析所有的试验据。如果数据能够成功证明药物的安全性和有效性,公司将向FDA提出新药申请。新药申请必须包括公司所掌握的一切相关科学信息。典型的新药申请有10万页甚至更多。根据法律FDA审核一份NDA的时限应该为6个月。但是几乎所有案例中的新药申请从首次提交到最终获得FDA批准的过程都超过了这个时限;在1992年对于新分子实体的新药申请平均审核时间为29.9个月。
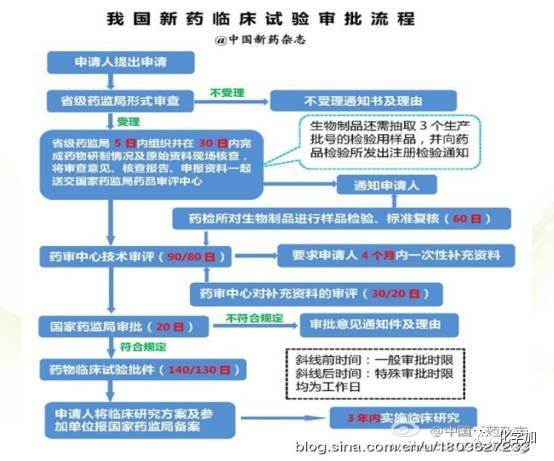
关于新药研发有一点不得不说一下,就是中国和美国在相关流程上有不同之处:
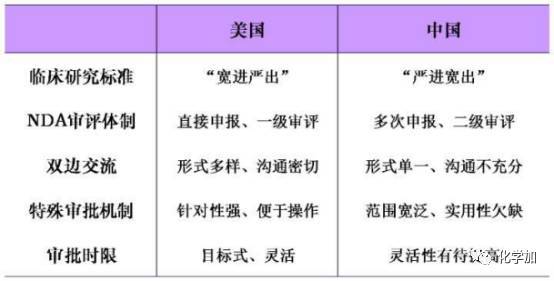
5、批准上市
一旦FDA批准新药申请后,该药物即可正式上市销售,供医生和病人选择。但是还必须定期向FDA呈交有关资料,包括该药物的副作用情况和质量管理记录。对于有些药物FDA还会要求做第四期临床试验,以观测其长期副作用情况。
1,部分图片来源于网络
2,《新药注册管理办法》、《CDER handbook》-FDA、《高通量筛选技术》、《新药临床前药理毒理评价申报 》、《药物临床试验基础知识》。
本文来源于化学加
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多