随着生物技术的飞速进步,生物药品在疾病治疗中的地位日益突出,已成为医药产业的重要增长点。然而,生物药的工艺开发和生产过程极其复杂,涉及多个学科和技术领域,对工艺水平和生产能力都提出了很高的要求。在此背景下,深入探讨生物制药工艺与生产的关键技术、发展趋势和面临的挑战,对推动我国生物医药产业的创新发展具有重要意义。
去年,珐成浩鑫特别设立了技术大讲堂这一学术品牌,主要聚焦生物制药,整合行业前沿理论与实操经验,搭建起知识与应用的高效转化桥梁,为生物制药产业升级注入强劲动力。值此2025年开年之际,PharmaTEC制药业联合PHARMAC珐成制药系统工程(上海)有限公司及其学术品牌珐成浩鑫技术大讲堂共同推出特别策划——《生物制药工艺与生产》专题,旨在为整个产业的高质量发展添砖加瓦。
此次专题内容汇集了众多来自生物制药行业的资深专家,他们将分享对生物制药工艺与生产领域的深刻见解和前沿思考。他们从“鑫” 视角出发,分享对生物制药工艺与生产领域的深刻见解和前沿思考,力求工艺精益求精。专题内容多元、覆盖面广,涉及生物制药工艺与生产的众多关键领域,我们将陆续刊登专业文章与君共赏。
生物制药设备的计算机化程度越来越高,必须有充分的验证来保证合规。依赖设备供应商提供的验证服务是不够的。本文结合生物制药设备的特点,分析了计算机化系统在运营阶段安全管理、备份与还原、定期回顾中常见的误区,并提出了相应的解决方案建议。

计算机化系统在制药设备中的应用日益广泛,特别是在生物制药的生产设备领域,例如配液、发酵、过滤、层析等环节都配备了计算机化系统。与传统制药设备相比,生物制药设备的计算机化系统有如下特点:
● 计算机化程度更高。
生物制药的生产设备几乎都配备了操作面板,并安装了各类软件,有些甚至内置了电脑。这些设备的运行实现了自动化控制,并能够记录相关数据;
● 计算机化程序的“黑箱”程度更高。
生物制药的生产设备从硬件及其复杂性来说,比传统制药设备有时显得更简洁,但其核心功能却在难以看见的软件部分。这些软件程序的运行逻辑是不可见的,只能通过“黑箱”测试来验证其一些显性功能。然而,很多隐性功能无法得到验证,这增加了用户深入了解设备的难度,并且在确保系统运行、验证以及法规符合性方面带来了额外的风险;
● 计算机化系统对供应商的依赖程度更高。
由于生物制药行业的兴起相对较晚,许多成熟或高端的生物制药设备均为进口产品。加之前述两个特点,生物制药工厂中熟悉生物制药设备的资深人员相对稀缺,他们往往无法独立起草验证文件,因此主要依赖设备供应商提供的验证文件。这种情况带来了一系列后续问题:不同设备厂家的验证思路和逻辑存在差异,这不仅导致了各设备厂家的验证文件质量的差异,还增加了合规性保证和运营管理的难度。
自中国 GMP 2010 版附录:计算化系统实施以来,国内药企在计算机化系统验证方面取得了显著的进步。然而,在计算机化系统验证中,普遍存在一种现象:重视开发阶段的验证工作,却忽视了运营阶段的管理。按照ISPE GAMP 5 的指导原则,一个完整的计算机化系统验证应该包括表 1 所列的内容(当然,基于计算机化系统的分类和复杂性,内容可以有适当删减,具体细节请参考 ISPE GAMP 5 的详细说明)。
综合前述生物制药的三大特点,生物制药工厂的计算机化系统验证在实际执行中,通常情况下是:项目开发阶段的验证测试由设备供应商负责。然而,设备制造商无法提供运营阶段的管理文件,因为运营阶段的管理文件是药企用户自身的文件,各药企的要求和书写格式各异,这就导致了运营阶段的验证管理存在不足。从计算机化系统审计的发现项数据来看,运营阶段的发现项数量通常多于项目开发阶段。
本文将对计算机化系统运营阶段的安全管理、备份与还原、定期回顾的常见误区进行分析,并从实际操作的角度提供一些建议。详细内容与具体要求可参考 ISPE GAMP 5 和 EUGMP 附录 11 以及其他相关法规。
误区一:
虽然工厂拥有一个总的计算机化系统安全管理标准操作程序(SOP),但缺乏针对各个计算机化系统的具体安全管理 SOP。总的计算机化系统安全 SOP 只能涵盖一些通用的安全要求和操作指导,例如,新购计算机化系统的安全权限配置要求、账号申请和注销的通用流程等。然而,各个具体的计算机化系统都具有其独特性。例如,同样是工程师权限,A 系统所具备的详细权限可能与 B 系统存在差异;再如,C 系统的重要性非常高,因此其安全管理的要求也需相应提高。就像维护管理一样,工厂虽然有总的维护管理 SOP,但是各个设备仍需拥有自己的维护 SOP 来明确其维护要求。同理,各个计算机化系统也应具备相应的安全管理 SOP,以规定本系统的安全要求。
误区二:
安全管理只考虑了账号与权限的分配管理。账号与权限的分配管理是安全管理 SOP 的核心部分,但一个完整的计算机化系统安全管理SOP 还应考虑以下内容:
● 账号与权限的其他方面:包括权限说明、临时用户的管理流程、员工调岗或离职时的账号处理、账号的退役流程以及密码的相关要求等;
● 物理安全:涉及操作站和数据存储区域的访问控制(如门禁系统或锁定设施)、防病毒软件、防钓鱼软件、防火墙等;
● 管理规定:对于系统权限配置的不合理性,如何通过 SOP 来规避风险。例如,某些工厂选择将系统管理员的职责统一规划给 IT 部门,本来的期望是系统管理员主要负责新建、调整、删除账号和权限分配,但是有些计算机化系统的系统管理员却具备执行所有操作功能的权限,而 IT 人员可能对系统的操作不熟悉,这导致 IT 人员作为系统管理员时存在较高的误操作风险,因此,合理的 SOP 规定对于降低此类风险至关重要。
表1 按照 ISPE GAMP 5的指导原则,一个完整的计算机化系统验证应涵盖的要素
误区一:
只备份了数据,而忽略了软件和参数的备份。备份通常涵盖三个主要方面:软件备份、参数备份以及数据备份。软件备份指的是对程序软件进行的备份操作,例如将软件存储在 U 盘中。计算机软件里一般包含一些设定参数,如 PID 调节参数、报警限设定等,这些参数已经过验证,应当进行备份,以便在执行灾难恢复时使用。这样可以避免灾难恢复后的再验证,或极大地简化验证流程。对于生物制药设备来说,由于前述计算机化系统的特性,这项备份尤其重要。此外,数据备份时容易遗漏审计追踪数据,这一点应在备份与还原的 SOP中明确指出。
误区二:
认为所有计算机化系统都需要备份其数据。这将导致备份的工作量和数据量成倍增加。正如计算机系统验证是为了符合法规要求,数据备份同样是为了确保符合法规规定。因此,必须明确数据是否用于 GMP 相关目的。对于那些用于 GMP 目的的数据,确实需要进行备份;而对于那些不涉及 GMP 的数据,则可以采取简化处理。通过这种方式,企业可以对数据是否用于 GMP 目的进行分类,具体分类标准参见表2。
表2 企业数据 GMP 用途分类标准
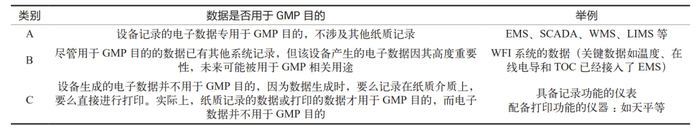
通过这种分类方法,A 类数据必须严格按照法规要求进行数据备份,而 C 类数据则无需进行数据备份(尽管企业可以选择备份,但此备份并非法规强制要求)。对于 B 类数据,企业可以基于自身情况来定义相应的备份策略。这种分类方法同样体现了风险管理在数据备份过程中的应用。
误区三:
备份与还原的 SOP 只关注备份与还原的具体操作步骤,却忽略了其他关键信息,例如人员职责、备份频率、备份保存期限等,这导致实际执行结果可能无法完全符合法规要求。一份完整的计算机化系统备份与还原 SOP 应当至少包括以下内容:
① 备份范围(明确哪些数据、参数和程序需定期备份);
误区一:
忽视账号的年度回顾。人员的工作变动和离职是正常现象,但随之而来的计算机系统账号与权限是否得到了及时的调整或删除(失活)。理论上,应该在人员工作变动或离职时及时进行账号调整或删除(失活),但不能完全排除存在疏漏的可能性,这就需要每年对所有计算机化系统里的账号与权限清单进行整理和重新审核,确保及时进行必要的调整或删除(失活),以避免系统里长期存在不合理的账号与权限分配。
误区二:
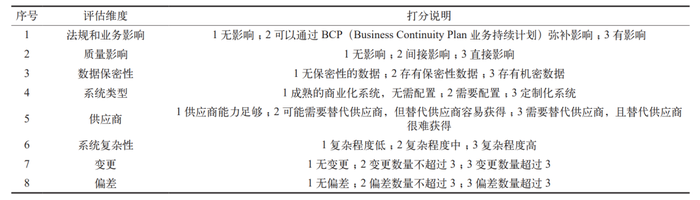
缺少定期回顾策略。很多企业在执行计算机化系统回顾时,常常面临一个难题:如何确定回顾周期?法规中并未提供明确答案。借助风险评估工具,通过表3 对所有需要定期回顾的计算机化系统进行评分,然后根据评分筛选出前 25% 的系统每年进行定期回顾,中间 50% 的系统每两年进行一次定期回顾,而最后 25% 的系统则每三年进行一次定期回顾。
接下来,列出一份计算机化系统的清单,按表3 评估出各系统的回顾周期,然后制定出年度的计算机化系统回顾计划,这是定期回顾策略文件的核心内容。定期回顾策略文件还应包含其他内容,例如回顾的范围、计算机化系统的回顾是否和所属设备的回顾合并等,本文不做阐述。
表3 计算机化系统定期回顾风险评估评分标准
鉴于生物制药设备的计算机化程度高以及数据的重要性,其计算机验证的难度也随之增加,审计的重视程度也会更高。因此,企业应该配备足够的专业人员,以确保计算机化系统的验证工作得到充分执行。
参考文献:
[1] 中国GMP 2010版附录:计算机化系统.
[2] ISPE GAMP5 第2版:A RiskBased Approach to Compliant GxPComputerized Systems.
[3] EU GMP Annex 11: ComputerizedSystems.

点击查看珐成浩鑫XBioR不锈钢生物反应器解决方案















评论
加载更多