我国高度重视仿制药的高质量发展,为进一步提升我国制药行业整体水平,保障药品安全性和有效性,陆续发布了《国务院关于改革药品医疗器械审评审批制度的意见》 (国发〔2015〕44号)、《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发〔2016〕8号)、《中共中央办公厅、国务院办公厅关于深化审评审批改革鼓励药品医疗器械创新的意见》,提出了开展仿制药质量和疗效一致性评价的总体要求。2020年5月14日,国家药品监督管理局发布关于开展化学药品注射剂仿制药质量和疗效一致性评价工作的公告,正式启动化学药品注射剂仿制药一致性评价工作,以期提升注射剂的整体质量和疗效,进一步推动我国医药行业转型升级和高质量发展。注射剂系指原料药物或与适宜的辅料制成的供注入体内的无菌制剂。注射剂可分为注射液、注射用无菌粉末与注射用浓溶液等。化学仿制药注射剂的过量灌装研究是药品研发、生产和审评过程中需要重点关注的问题,为进一步明确过量灌装的技术要求,国家药品监督管理局药品审评中心于2024年7月5日发布《化学仿制药注射剂过量灌装研究技术指导原则》,为相关制剂研发、生产过程中可能存在的过量灌装问题提供了指导思想和研究思路[1]。笔者从该指导原则相关背景和技术要求入手,结合审评中遇到的过量灌装实例进行探讨。本文仅针对临床使用时需要转移的注射剂进行讨论,不涉及大容量注射液和预灌封注射剂等。
注射剂的过量灌装(overfill)是指每个容器中药品的灌装量较标示量适当增加的体积或重量,以确保实际给药剂量满足说明书中规定的用量。
与口服固体制剂直接服用不同,注射剂在临床使用时,通常需要通过中介设备(例如注射器)或与基础输液配伍后使用,因注射器很难或不可能从包材中取出100%的内容物,即包材粘附损失等原因,导致注射剂实际装量与最终使用到人体的药量往往有一定差异,致使给药剂量不足,因此需要过量灌装。
研究中应注意区分“过量灌装”与“过量投料”。根据ICH Q8(R2),过量投料(overage)是指为补偿预期的并被证明了的生产中的损失,在生产过程中原料药的实际投料量超过了处方要求。一般不提倡在药品生产中过量加入原料药,以补偿在生产或者货架期内的降解,或试图延长产品的货架期[2]。
美国食品药品监督管理局(FDA)于2015年6月发布的指南文件《Allowable Excess Volume and Labeled Vial Fill Size in Injectable Drug and Biological Products》显示,灌装量过大可能会导致用药错误,滥用剩余药物产品或合并小瓶以获得单次剂量[3]。剂量的合并或单个药瓶的重复使用会增加患者间血源性疾病传播的风险,尤其是对于价格昂贵的化疗药物,若医护人员收集多个过量的剩余药物,重新组合成一支完整的药剂给患者使用,患者的不良事件暴露率将大大增加,最明显的是微生物污染引起的事件。因此,FDA建议在没有适当理由的情况下,减少小瓶的过量灌装,以期减少药物浪费、用药错误、不良事件和液体药物产品的滥用。
FDA于2022年 发 布 的《Allowable Excess Volume/Content in Injectable Drug and Biological Products》Manual of Policy and Procedures(以下简称MAPP文件)显示,若小瓶中的内容物不足以完成标示剂量的递送,那么医护人员或消费者则不得不使用不足的剂量进行递送或使用额外的小瓶完成标示剂量的递送。灌装容量不足会在患者给药期间引起问题[4]。
由此可见,“过量灌装”研究在化学仿制药注射剂研究中尤为重要。因此,在研发阶段,应充分考虑灌装量的大小对临床疗效及安全性的影响,以规范注射剂研发及申报工作。本文将从国内外监管机构的要求、过量灌装计算方法以及典型案例分析等方面对“过量灌装”问题加以阐述。
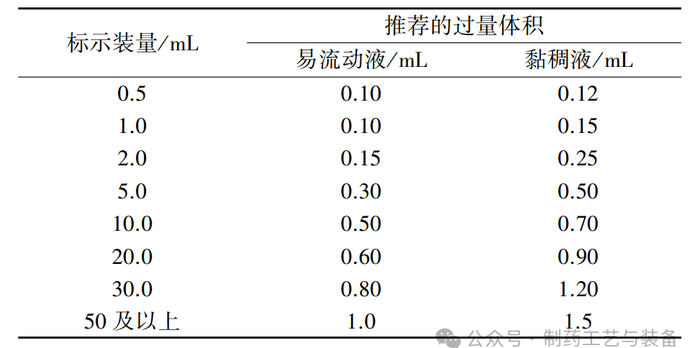
由于产品的黏度、表面张力、小瓶尺寸、剂量和其他因素,不同产品之间过量灌装量可能会有所不同。近年来随着对药品研究的不断深入,国际组织和各国药品监管机构也逐步加深对药物过量灌装的理解和认识,逐步明确了过量灌装的技术要求。《美国药典》注射剂通则 USP<1>[5]、<697>[6] 指出,每个注射剂容器的灌装体积应略超过标签的标示量,增加的装量需满足可抽提体积(见表1),确保每支(瓶)产品的可转移剂量均不低于标示量,以满足注射剂临床疗效及安全性需求。《美国药典》 <1151>[7] 推荐的不同装量的易流动液和黏稠液体建议的过量灌装体积,具体见表2,通常<1151>推荐的过量体积足以满足可抽取和给药的标示体积。美国 FDA 发布的 MAPP文件指出注射剂标签上的净含量系指最小剂量,以符合标签要求和可接受的过量灌装量,需增加的灌装量应遵照 USP 通则执行,但为避免临床用药困惑,过量灌装量通常不需要在标签和说明书中特别说明。仿制药的灌装量应基于药物的临床使用习惯、药物的治疗窗、国内外药典以及其他文献综合考虑,同时 FDA 也不会针对过量灌装提出一个严格的框架来对所有药品进行一刀切的监管。
表1 国内外药典装量检测方法对比
《英国药典》虽未明确过量灌装的定义,但肠外制剂通则显示注射剂容器中的液体体积应满足标示剂量可抽取体积和需注射的体积(2.9.17)。
这些技术文件大幅提升了过量灌装研发的准确度,可用于指导对药物过量灌装合理性的监管。
《中国药典》2020 年版 0102 注射剂项下规定了注射剂的灌装标示装量不大于 50 mL 时,可适当增加装量,为药物装量的检测和允许增加的装量提供了参考。《中国药典》推荐的增加量与《美国药典》,与上文表2 基本一致,主要差异是《中国药典》未规定标示装量为 30mL 时,推荐的过量体积。
装量检查用于确认和检测制剂的灌装量,以保证其在使用时用量不少于标示量,以达到临床用药剂量的要求。
对于注射液及注射用浓溶液照《中国药典》2020 年版(四部)0102 注射剂项下【装量】项下的方法进行检查,即为可抽取体积;对于注射用无菌粉末,建议研究可抽取体积和总药量,根据参比制剂说明书的用法,加入说明书中规定的溶剂将样品进行复溶,参考《中国药典》检测出样品的可抽取体积,药液浓度乘以可抽取体积,即为可抽取总药量(见表2)。绝对体积可采用重量除以相对密度计算装量,操作如下:取供试品 5 个(50 mL 以上者 3 个),除去外盖和标签,容器外壁用适宜的方法清洁并干燥,分别精密称定重量,除去内容物,容器用适宜的溶剂洗净并干燥,再分别精密称定空容器的重量,求出每个容器内容物的重量与平均重量,采用平均重量除以相对密度计算绝对体积。国家药典委发布的《中国药典》通则 0102 注射剂(征求意见稿),显示修订后的装量检查方法与现行《美国药典》相似,后续以正式稿为准[8]。增加的灌装量包含使用时注射器的损失(黏附及针头的保留量,使用时不可能将针头内的量也注尽),因此在倾至量具时《美国药典》规定不排尽针头中的液体。
《化学仿制药注射剂过量灌装研究技术指导原则》明确了仿制药过量灌装的研究思路、研究内容及关注点、申报资料要求等。该指导原则对注射液和注射用无菌粉末过量灌装计算方式进行了说明。
对于注射液,申请人可按下式计算灌装量范围:灌装量下限=标示装量+过量体积(mL),灌装量上限=标示装量×合理论证的系数+过量体积(mL)。申请人在确定灌装量上限时,应对系数进行合理论证,可能的考虑因素包括但不限于灌装精度、产品特点等。例如,某注射剂标示量为 5 mL,根据《中国药典》规定可增加 0.30 mL 的装量,则灌装量下限为 5.3 mL;假设合理论证的系数为 1.1,则灌装量上限为 5.8 mL。
对于注射用无菌粉末,参比制剂说明书中明确复溶后浓度的情形,申请人可按下式计算灌装量范围:总药量下限 = 复溶后标示浓度(mg·mL-1)×复溶后总体积,总药量上限 = 复溶后标示浓度(mg·mL-1) ×合理论证的系数×复溶后总体积。FDA 培 训 资 料 《 MAPP5 019.1 Allowable Excess Volume/Content in Injectable Drug and Biological Products》进行了举例说明:对于400mg 规格(净含量)单剂量冻干粉针剂,用 20 mL 注射用水复溶后,得到复溶后浓度为 20 mg·mL-1的溶液。复溶后体积=20 mL 注射用水+1.4 mL 膨胀体积(由于粉末溶解,药液体积比加入的复溶液体积“膨胀”的部分) = 21.4 mL ( 备注: 1.4 mL 过量灌装符合 USP<1151>的要求)。为达到标示浓度需每瓶加入的活性成分(API)量=总药量下限=100%复溶后标示浓度×复溶后体积=20 mg·mL-1×21.4 mL = 428 mg;总药量上限=110%复溶后标示浓度×复溶后体积 =22mg·mL-1×21.4 mL = 471 mg。若灌装液理论浓度为40 mg·mL-1,实际测量浓度为39.6 mg·mL-1(99%),则灌装量下限=428 mg÷39.6 mg·mL-1= 10.8 mL;灌装量上限 = 471 mg÷39.6 mg·mL-1 = 11.9 mL。若按照 97% ~103%的范围,则灌装量下限 = 428 mg÷(40mg·mL-1 ×97%) = 11.0 mL;灌装量上限 = 471 mg÷(40 mg·mL-1×103%)= 11.4 mL[9]。
对于注射用无菌粉末,参比制剂说明书中未明确复溶后浓度的情形,仿制药研发时可根据参比制剂说明书的用法进行复溶后,仿制药的可转移剂量下限应满足说明书标示的给药剂量,同时上限应避免过度过量,拟定的过量灌装范围需结合参比制剂说明书提供充分的支持性研究资料。
在审评实践中,采用冻干工艺生产的注射用无菌粉末的过量灌装问题较为常见,且较多品种由于该问题而导致注册失败。下面结合过量灌装指导原则,以此类冻干制剂的典型产品为例,探讨注册审评中过量灌装研究的常见问题及考量。
情形一:参比制剂明确了复溶溶液的浓度。欧洲EMA公开资料显示,每瓶注射用伏立康唑Vfend®(规格 0.2 g)过量灌装 1.13 mL(约5.65%),以便复溶时可提取体积为 20 mL,相当于 200 mg 伏立康唑;在用 19 mL 注射用水复溶后,所得溶液中每1 mL 含有 10 mg 伏立康唑[10]。美国 FDA 说明书明确了注射用利福平 Rifadin®IV 加 10 mL 灭菌注射用水后的复溶溶液浓度为 60 mg·mL-1 [11]。注射用阿奇霉素原研进口产品Zithromax®500 mg 规格的说明书显示,制备药品原液时,向本品瓶中加4.8 mL灭菌注射用水,振荡直至药物完全溶解,使每毫升溶液中含 100 mg 阿奇霉素[12]。这 3 个品种均属于指导原则中参比制剂说明书中已明确复溶后浓度的情形,申请人应参照指导原则开展过量灌装研究。
情形二:参比制剂明确了过量灌装量。注射用达托霉素 Cubicin 的 EMA 审评报告指出,本品过量灌装5%,以确保在复溶后能够从瓶中抽出产品标示量的达托霉素。0. 35 g 和 0. 5 g 规格过量灌装量均为5%[13]。注射用磷酸特地唑胺(规格 200 mg)原研药品专利 CN102439006A 和日本上市说明书 IF 文件均明确原研药品过量灌装 5%,200 mg规格注射用磷酸特地唑胺的实际装量 210 mg,以抵消临床转移时的损失[14-15]。这两个品种公开的参比制剂过量灌装信息较明确,仿制药可参考其进行过量灌装研究。同时提醒仿制药企业关注,在确定仿制药的过量灌装量时,因包材、工艺、测试精度等差异,不建议仿制药机械套用参比制剂的过量灌装数据,而应结合自身生产实际情况、说明书要求、参比制剂信息,经充分合理的评估后,拟定合理的过量灌装量。若确实需拟定与参比制剂不一致的参数,应提供合理性论证,且应确保每支(瓶)产品的实际给药剂量均不低于标示量。
情形三:参比制剂说明书未明确复溶后溶液的浓度,且无过量灌装的公开信息。如,注射用替考拉宁。对于此类产品,仿制药企业应对参比制剂的过量灌装量进行规范的剖析,按照《化学仿制药注射剂过量灌装研究技术指导原则》对仿制药拟定合理的过量灌装量。
过量灌装指导原则指出,化学仿制药注射剂的过量灌装宜与参比制剂保持一致,如不一致需提供合理性论证,应确保每支(瓶)产品的实际给药剂量均不低于标示量。该指导原则指出仿制制剂可拟定与参比制剂不同的过量灌装量,但需要提供充足的合理性论证。但如果参比制剂存在过量灌装,而仿制制剂未过量灌装,通常是不被认可的。FDA 发布的行业指南《ANDA Submissions-Refuse-to-Receive Standards Guidance for Industry》显示,若注射剂过量灌装量与参比制剂不一致,且超出药典有关药品专论中建议的过量灌装范围,应事先经适宜性请愿程序批准,否则会拒绝受理(Refuse-to-Receive)[16]。因此仿制药开发时,应严格测定参比制剂过量灌装的量,当过量灌装的量不一致时,应提供充分合理的论证。审评实践中,部分申请人因参比制剂选择不合理、参比制剂参考文献调研不充分,或仅对极少批次参比制剂含量进行检测,从而确定错误的参比制剂过量灌装量,对仿制药开发和注册将产生不利影响。有些申请人测定参比制剂过量灌装量较低,如2%、3%时,会错误地认为参比制剂未进行过量灌装,据此设计的仿制药的灌装量与参比制剂存在差异,注册审评中将导致不批准的情况。建议研发阶段充分调研产品信息,包括相关专利、文献、公开的审评报告、说明书等,并对多批参比制剂进行详细研究,综合分析参比制剂的过量灌装情况,选取的参比制剂的批次数量和每批参比制剂抽取的样本数量均应具有统计学意义并能代表参比制剂的质量,以消除信息不准确、批间差异等影响。
质量标准中含量限度是保障药品质量的关键指标,研究人员应根据产品特性、过量灌装指导原则、参比制剂信息、稳定性数据等,拟定合理的含量限度。根据含量计算方式,过量灌装量对注射液的含量没有影响,但对于冻干粉针剂,其含量计算基于平均装量,因此,含量限度应根据过量灌装情况而上移。建议含量限度同药典标准或国家标准一致,若经研究上述限度确实无法满足需要,则可适当调整含量限度范围,并提供限度制定依据。必要时,待产品批准后根据相关要求,联系药典委修订国家标准。同时建议加强医护人员宣贯,严格按照说明书用法用量给药,避免因给药剂量增大而引发安全性风险。
药液的性质、灌装体积、灌装速度等均可能会影响灌装量精度,因此良好的生产过程控制是确保过量灌装准确性的关键。FDA 建议进行100%的在线灌装重量检查,或者拟定合理的取样计划,如至少在灌装工序的开始、中间和结束阶段对每个填充针进行装量差异检查。仿制药生产企业可根据自身生产条件拟定合理的取样计划,并在工艺验证时进行相关研究,灌装量应被严格控制在规定的装量范围内,且应整个生产期间灌装量均一性,不应出现明显的上升或下降趋势。
通常注射剂过量灌装会对原辅料用量、灌装时间等产生影响。值得注意的是,对于冻干制剂,增加灌装量通常需要重新开展冻干工艺研究,以确保增加灌装量前后的冻干制剂质量一致。
因生产线不同、包材成本、新包装材料和技术的应用等原因,在确保仿制药质量和稳定性不低于参比制剂的基础上,仿制药生产厂家最终确定的内包材材质可能与参比制剂有所区别,而不同材质对注射液的黏附体积存在差异,需逐案分析。但总原则应确保每一瓶(支) 的实际给药量均不低于说明书标示的给药剂量。
过量灌装是化药注射剂仿制药研发、生产和注册审评中需要重点关注的内容,涉及药物的生产控制和管理等领域,其最终的目的是保证临床给药剂量的准确性和合理用药。《化学仿制药注射剂过量灌装研究技术指导原则》的出台,为我国注射剂过量灌装研究提供了技术支撑,我国对注射剂开发的监管也逐渐完善。本文概述了过量灌装问题产生原因及风险、国内外药品监管机构对过量灌装研究的技术要求,并根据过量灌装指导原则对测定方法、典型案例、常见问题做了剖析,旨在引导读者不断加深质量源于设计(QbD)的理念,在研发早期对药物过量灌装进行详细研究和分析,降低开发风险,并在今后的研发和生产实践中,不断加强对药物制备工艺细节的认识和管控,为向临床提供质量更高、安全性更好、疗效更显著的药品而不断努力。
[1] 国家药品监督管理局药品审评中心.化学仿制药注射剂过量灌装研究技术指导原则 [EB/ OL].(2024-07-05) [ 2024 - 08 - 01 ]. http: / / assist1. cdeapp. org. cn /office / homeForeignGuide / toSearchModel.
[2] ICH.ICH Q8(R2)Pharmaceutical development[EB/ OL].(2009- 08 - 01) [ 2024 - 08 - 01]. https: / / database. ich.org / sites/ default / files/ Q8%28R2%29%20Guideline.pdf.
[3] FDA.Allowable Excess Volume and Labeled Vial Fill Sizein Injectable Drug and Biological Products [ EB/ OL].(2015-05) [2024-08-01].https: / / www.fda.gov / regulatory-information / search-fda-guidance -documents/ allowable-excess-volume-and-labeled-vial-fill-size-injectable-drug-and-biological-products.
[4] FDA.Allowable Excess Volume / Content in InjectableDrug and Biological Products.[EB/ OL].(2022-01-28)[2024 - 08 - 16]. https: / / www. fda. gov / media / 155066 /download.
[5] U. S. Pharmacopeia Convention. Injections and ImplantedDrug Products ( Parenterals )—Product Quality Tests[S].(2024- 05- 01) [2024- 08- 01]. https: / / doi. org /10.31003 / USPNF_M98730_04_01.
[6] U.S.Pharmacopeia Convention.Container content for injections[S]. ( 2015- 05- 01) [ 2024- 08- 01]. https: / / doi.org / 10.31003 / USPNF_M7620_01_01.
[7] U.S.Pharmacopeia Convention. Pharmaceutical dosageforms[ S]. [ 2024 - 08 - 01]. https: / / doi. org / 10. 31003 /USPNF_M99860_10_01.
[8] 国家药典委员会.关于征求«中国药典»通则(0102)注射剂修订草案意见的通知[EB/ OL]. ( 2023- 06- 25)[2024-07-20].https: / / www. chp. org. cn / # / newsDetail?id = 17843.
[9] FDA.MAPP 5019.1 Allowable Excess Volume / Content inInjectable Drug and Biological Products[EB/ OL].(2022-09-20) [2024-07-20]. https: / / www.fda. gov / media /166583 / download.
[10] EMA. Vfend: - EPAR Scientific Discussion [ EB/ OL].[2024- 07 - 20]. https: / / www. ema. europa. eu / en / documents/ scientific-discussion / vfend-epar-scientific-discussion_en.pdf.
[11] FDA.Rifadin[EB/ OL].(2023-12-08) [2024-07-20].https: / / www. accessdata. fda. gov / drugsatfda _ docs/ label /2023 / 050420s091,050627s036lbl.pdf.
[12] EMA.Zithromax[ EB/ OL]. ( 2021 - 11 - 22) [ 2024 - 07 -20]. https: / / www. accessdata. fda. gov / drugsatfda _ docs/label / 2021 / 050733s050lbl.pdf.
[13] EMA.CUBICIN:-EPAR Scientific Discussion[EB/ OL].(2006- 11 - 08 ) [ 2024 - 07 - 20 ]. https: / / www. ema.europa. eu / en / documents/ scientific - discussion / cubicin -epar-scientific-discussion_en.pdf.
[14] 道格拉斯·菲利普森,卡塔琳娜·赖兴贝歇尔,罗伯特·J·杜吉德,等.(R)-3-(4-(2-(2-甲基四唑-5-基)吡啶-5-基)-3-氟苯基) -5-羟甲基噁唑烷-2-酮二氢磷酸酯的晶型:102439006A[P].2002-05-02.
[15] PMDA.注射用磷酸特地唑胺 IF 文件[EB/ OL].(2021-07-06)[2024-07-20].https:/ / www.pmda.go.jp/ PmdaSearch/iyakuDetail/ ResultDataSetPDF/ 170050_6249402D1026_2_02.
[16] FDA.ANDA Submissions Refuse -to -Receive StandardsGuidance for Industry [EB/ OL].(2016-12)] [2024-07- 20 ]. https: / / www. fda. gov / regulatory - information /search - fda - guidance - documents/ anda - submissions -refuse-receive-standards-rev2.














评论
加载更多