非最终灭菌无菌制剂的污染控制策略
2022 年 8 月 22 日,欧盟正式发布了 EU GMP 附录 1《无菌药品生产》,其强调应在整个设施内实施污染控制策略(Contamination Control Strategy,CCS)以确定所有关键控制点,并评估用于管理医药产品质量和安全风险的所有控制(设计、程序、技术和组织机构)和监测措施的有效性。应积极审核并酌情更新 CCS,并应推动生产和控制方法的持续改进。本文从人、机、料、法、环全方位出发,用思维导图梳理了产品污染的潜在风险,为实际工作中 CCS 的撰写提供了参考。
制定 CCS 需要专业的技术和工艺知识,潜在的污染源可归结为微生物和细胞碎片(如热源、内毒素)以及颗粒物(如玻璃和其他可见异物和不溶性微粒)。欧盟在 GMP 被广泛接受和实施的情况下再强调 CCS,说明 GMP 及各种监管法规并不能够完整地覆盖 CCS。所以在撰写 CCS 的时候,不能够仅束缚于现有的监管法规,而是应该采用头脑风暴来拓展思路,充分挖掘出污染风险的潜在因素。下文将展示如何用思维导图来梳理人、机、料、法、环各个方面存在的产品污染的潜在风险。
即便采用的是全自动无人操作的药品生产线,设计人员的法规意识仍有可能会间接影响到其产品质量。所以对于人员在线操作的药品生产线来说,人员更是与产品的质量密切相关。常言道,人员是生产过程中最大的污染源,因此也需要对于操作人员进行屏障隔离,以免操作人员本身产生对药品的污染,以及高活性药物生产时对操作人员的影响。
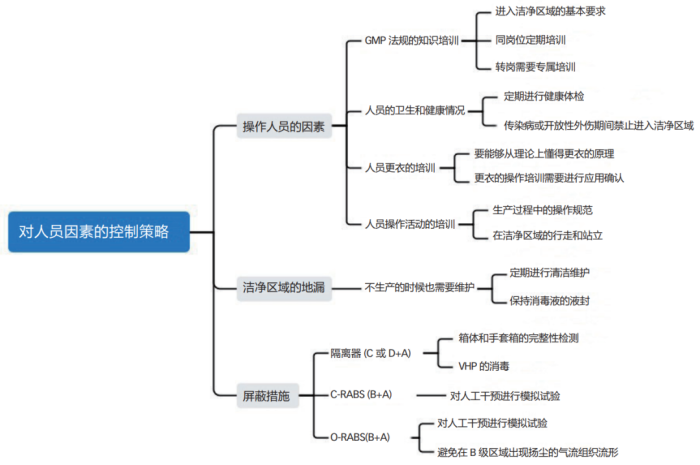
可以用如图 1 所示的思维导图来梳理一下人员方面造成产品污染的潜在因素,这样就便于采取措施来应对和化解风险,乃至于达到提前消除风险的目标。其中,屏蔽措施虽然讲的是设备,但主要涉及到对人员的屏蔽,所以就将其安排在此处一并讨论。
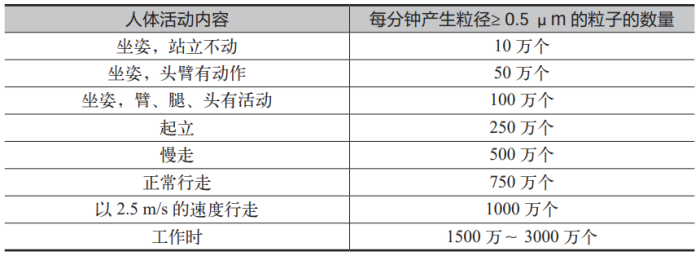
在图 1 的思维导图中,涉及到的内容基本都是药企容易理解并做到的,但还是有几个方面需要特别提醒一下。据统计,洁净室中空气的发尘量占总体的 5% ~ 10%,装修的发尘量占总体的20% ~ 30%,气体和液体的发尘量占总体的 5% ~ 10%,机器的发尘量占总体的 20% ~ 30%,人员的发尘量占总体的占 30% ~ 40%,由此可见人是洁净室中最大的污染源;而且人体散发出来的粒子数量,还与人体行为和肢体幅度有关,详见表 1。所以操作人员在洁净区域的肢体运动要尽量地迟缓,特别是在B 级区域的操作活动时更应该注意。在RABS 的 B 级背景房间内,如果暂时没有操作活动,需要人员尽量站立在下风侧的回风口附近,但还不能阻挡回风通道,从而最大限度地减少人员对环境的负面影响。允许进入洁净区域的人员数量,也需要与设计时的换气次数相适应。
另外,传统设计的 O-RABS(B+A)系统,一般都是从 B 级房间的送风口取风的,这就埋下了一个不可避免的隐患。由于 RABS 系统内是层流,所以需要的风量较大,而 B 级区域送风口的一次风满足不了 O-RABS 层流的需求量。因此O-RABS 系统会将其下部排出的风再次从上部吸入,在 B 级背景房间的局部区域引起气流由下往上的扬尘流形,这会将操作人员身体上脱落的颗粒物扬起。要避免这一弊端,O-RABS 系统需要直接通过静压箱来送风,而不再与B级背景房间抢风。
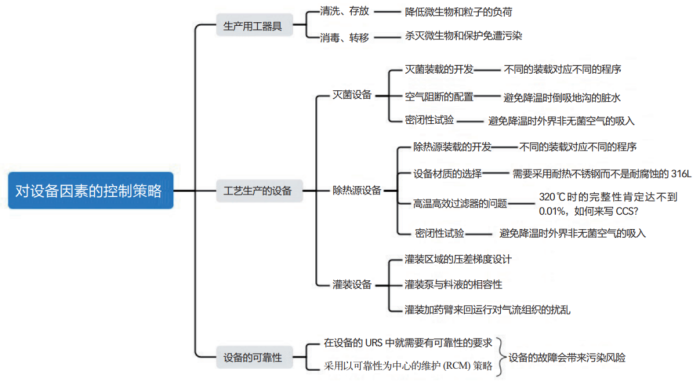
生产设备可以再细分为生产用的工器具、工艺生产设备和公用工程设备,同样也可以用思维导图来梳理一下其污染控制的策略(如图 2 所示)。
生产用的工器具有些是直接与物料相接触的,所以在材质的选用上必须采用 316L 的不锈钢。与物料相接触的工器具则需要灭菌和除热源。干热除热源的过程自然也能起到灭菌的作用,但干热除热源的温度比较高,甚至会达到 320℃的温度,这么高的温度并不是普通的316L 所能耐受的,所以不建议采用干热灭菌柜来除热源。按照 FDA 的规定和新版 GMP 指南中的要求,可以用无热源的注射用水(WFI)淋洗工器具以除去热源,然后仅用湿热灭菌柜来进行灭菌处理。
虽然上文提到尽量不使用干热灭菌柜来除热源,但在生产无菌注射剂的时候还必须使用隧道烘箱来除热源,这就给 CCS 带来了很大的麻烦。高效过滤器只能够在室温的状态进行完整性检测,从理论上讲温度上升后泄漏程度会增加。如果在隧道烘箱加热段的高温条件下,高效过滤器在工作时发生泄漏,这虽然不会引起无菌和热源的问题,但必然会造成一定程度的粒子污染,最终会影响到可见异物和不溶性微粒的质量指标。目前,该污染虽然仍是无法完全避免的,但也不能够放任自由、听天由命,药企需要对其污染的程度进行了解和掌握。由于篇幅的关系在此不详细展开,读者们可以根据自己的实际情况进一步研究。
湿热灭菌设备在完成灭菌后需要进行冷却,在冷却的过程中箱内会产生负压,这样便可能会有从外界下水道吸入脏水和室内非无菌空气倒灌的情况发生。按照 GMP 的相关规定,防止下水道脏水的倒灌需要配置空气阻断。为了防止室内非无菌空气的倒灌,就需要定期地对设备进行负压检漏,以达到一定的密闭程度。对于设备放空使用的无菌过滤器,也需要定期进行完整性的检测。
另外,需要引起注意的一点是设备的故障,其本身或许不产生污染,但在维修的过程中可能会导致污染的发生,所以需要在编写设备选择的用户需求说明(URS)开始便关注其可靠性。国际制药工程协会(ISPE)在 2020 年就颁布了设备的可靠性指南,在我国 2023年新版的 GMP 指南中也提出了以可靠性为中心的维修(RCM)的概念。药企需要在维护的过程中利用 RCM 的理念,提高设备的可靠性,避免设备在生产过程中发生故障。
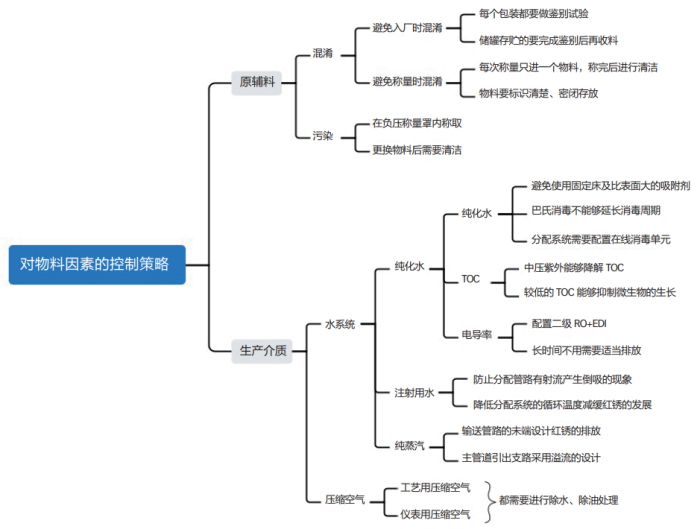
物料的污染控制需要从源头开始。历史上,在生产物料混淆方面曾发生过一起惨痛的事故:不法供应商将二甘醇冒充药用辅料丙二醇出售给制药厂,致使假冒辅料投料用于生产,制造出假药“亮菌甲素注射液”并投入市场,最终酿成了极其严重的后果。所以在原辅料入厂的时候,必须严格把关,对每件原辅料都做好鉴别。对物料方面的风险采用思维导图梳理,如图 3 所示。
图3 物料因素的污染风险分析
对于多组份的复方制剂,在配料的时候需要额外地予以关注,如果流程设计不合理就容易导致混淆或者称配错误。
生产介质也是直接与原辅料混合的,而且它的用量一般相对于原辅料来讲还比较大,所以介质的质量控制至关重要。其中,纯化水既是工艺介质又是基础原料,注射用水和纯蒸汽则是由纯化水制备获得的。注射用水主要用于关键区域的卫生和药物的配料,纯蒸汽主要用于设备的直接消毒,加湿用的纯蒸汽会弥漫在整个洁净区域内。
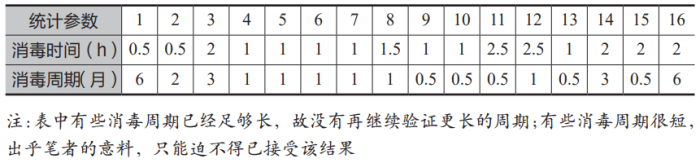
纯化水中的微生物在蒸馏时能被高温杀灭,但水中的革兰氏阴性菌在杀灭后会释放出内毒素。由于纯化水的微生物检测是滞后的,因此纯化水中微生物的控制尤为重要。笔者曾经调研了国内 14 家药企的 16 套纯化水制备系统,表 2 列出了这些系统的消毒周期和巴氏消毒的时间。
表2 调研 16 套纯化水系统的消毒数据
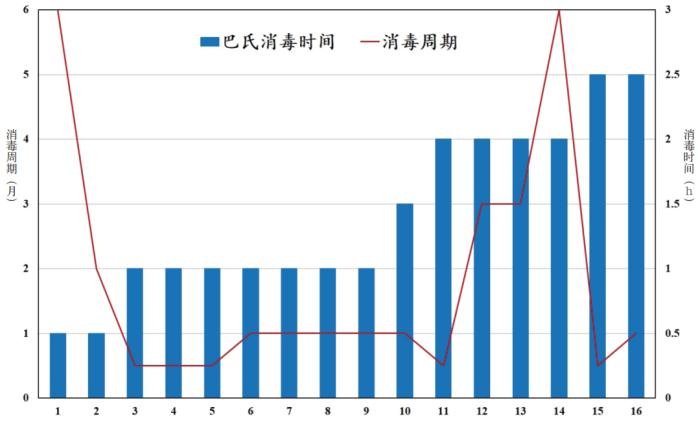
将表 2 中的数据作图得到如图 4所示的曲线,从图 4 中可以清晰地看到,巴氏消毒所维持时间的长短,与系统消毒周期的长短没有明确的关联性。但有一点是可以肯定的:采用全膜法制备纯化水的工艺,其微生物的控制效果远远好于传统的活性炭工艺。而且对于规模大一点的情况来说,全膜法的投资成本还低于活性炭工艺。
EU GMP 附录 1《无菌药品生产》中还提到,注射用水循环分配系统的温度达 70℃即可。所以在实际的运行中,只要控制温度≥ 75℃就可以了,没有必要维持在 80℃以上,否则既会增加能源成本还容易招致红锈的产生。
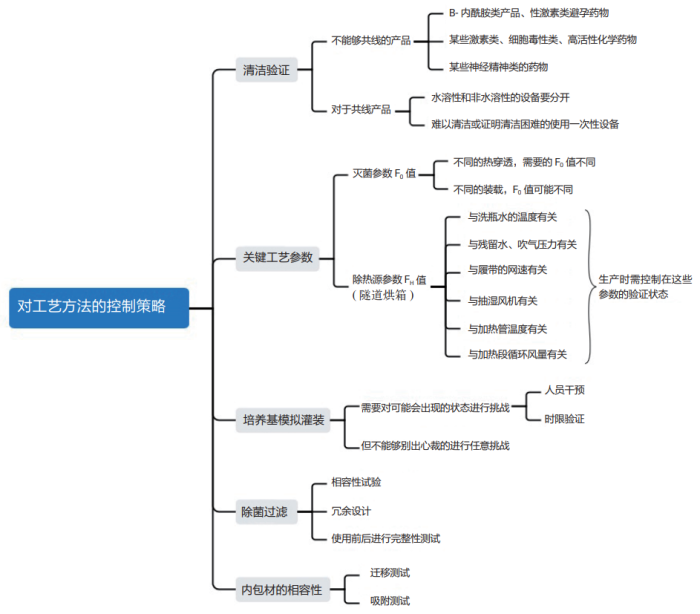
工艺方法方面主要会涉及到清洁验证和工艺控制参数,同样也可以用思维导图进行梳理,如图 5 所示。有些国家明文规定需要采用独立的空调系统,那就不能够用验证来说明可以共用空调系统。对于培养基模拟灌装试验来说,需要模拟诸如人工干预等情况,但也不能够夸张地来模拟试验一些不符合实际的动作和情形。
除菌过滤的温度、压力和时限是通过验证确认的,最终的除菌过滤器应当尽可能接近灌装点,即尽可能地省略除菌过滤后料液的中间储存环节。如果除菌过滤装置是离线灭菌的,那么在装配的时候需要配置无菌连接器。
尚在产品研发阶段,就需要对除菌滤芯进行细菌截留试验、化学兼容性试验、可提取物或浸出物试验、安全性评估和吸附评估等研究。对过滤过程中潜在可能引入的杂质和风险进行充分的评估。
同样,内包材与药物在产品研发时也需要进行相容性试验,胶塞的基材有氯化和溴化两种,一旦不兼容便会出现浊度不合格的情况,那就要使用覆膜或者镀膜的胶塞。
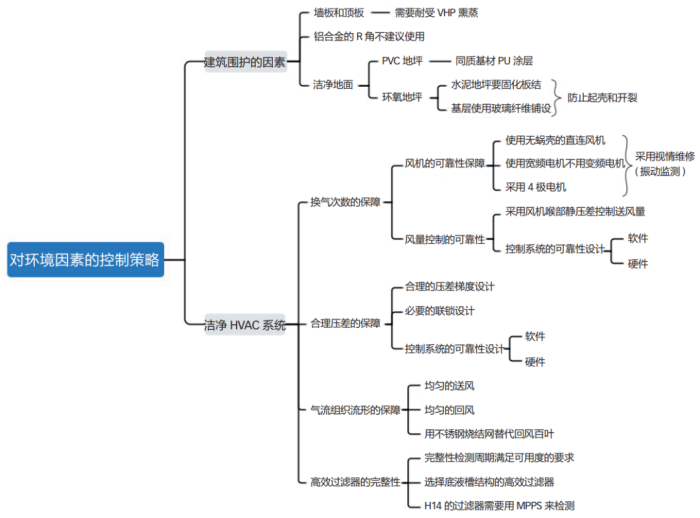
同样,使用思维导图对环境因素中的污染风险进行梳理分析,如图 6 所示。药品生产的洁净环境是保证产品质量的基本条件,除尘最终是依靠高效过滤器来完成的。特别是对于非最终灭菌的注射剂来说,高效过滤器的完整性是产品质量的关键因素。所谓的洁净区域是具备自净能力的,也就是说随着时间的推移会越来越干净。然而有些企业似乎每间隔一段时间,必须对区域进行一次熏蒸来杀灭微生物。这就说明了洁净系统存在着问题,这有可能是高效过滤器泄漏,也有可能是系统设计存在“bug”——自净存在死区。
有些药厂的高效过滤器完整性数据,离奇得好,甚至于超过供应商标准一个数量级。数据高于标准一个数量级的高效过滤器可能并没有问题,但是此时在同一系统中数据没有高出一个数量级的高效过滤器很可能是有问题的,是个人检测与标准的检测间存在一个系统偏差,这个偏差可能来自于检测器的类型、气溶胶的粒径。在标准 EN 1822-42009 第 40 页的 AnnexE.4 章节“Leak criteria”(泄漏标准)中有这样的描述:“对于 H13 级别过滤器(整体 MPPS 效率大于 99.95%,局部 MPPS 效率大于 99.75%)来说,对0.3 ~ 0.5μm 粒径粒子的效率必须大于 99.9996%”,可想而知粒径对效率的影响有多大。
房间的气流组织流形比换气次数更加重要,如果做不到四面回风,至少也要保证有两侧的回风。房间内适度的高压对于避免交叉污染是有利的,但压力越高滞留房间的气体就越多,这样所造成的返流情况就会越明显,反而会影响到房间的自净效果。所以在设计房间压力的时候,没必要盲目追求很高的压力,压力过大还会影响建筑围护的气密性。
在有物理隔断的时候,压差能够避免交叉污染的发生。一旦人员进出时门被打开,这个静压差便会陡然地降低。不能毫无理论依据地规定允许延时多长时间,门打开后需要能够维持 20 cm/s的风速,这样也能够避免交叉污染的发生,这在暖通泰斗许钟麟先生的教科书中是有描述的。
有些药企会模拟临时停电,验证来电后经过清洁卫生并自净一段时间后即可恢复生产。这对于 B+A 的区域来说风险是很大的,因为验证是将系统作为“黑箱”来试验的,其中存在很多的偶然情况。需要将系统作为“白箱”来试验,这样的验证才有实际意义。
另 外, 如 果 采 用 的 是 RABS 的B+A 系统,在洗瓶间和灌装间的入口需要设置两个等压的缓冲室;否则在隧道烘箱运行的时候,洗瓶间或(和)灌装间人员进出的开门,将严重影响到隧道烘箱内的压差平衡,从而影响到隧道烘箱的 FH 值和除热源的效果。关于这点可以参考国家实用新型专利CN203550481U“一种用于维持隧道烘箱两端压差稳定的系统”。
法规要求积极审核并酌情更新CCS,所以在每年一度的质量回顾中,需要对关键质量数据进行统计学分析。画出这些关键质量数据的控制图,计算出其工序能力指数,来发现 CCS 的薄弱环节甚至于是盲区。如上述提到的隧道烘箱高温高效问题,就需要药企对每一批次产品的特定参数进行统计学分析,以掌握其高温高效的泄漏程度和污染风险。
可以借助于 PDCA 循环的模型,持续不断重复这个质量改进循环,达到降低产品风险、提高产品质量的最终目的。
撰稿人 | 钱杨华
责任编辑 | 邵丽竹
审核人 | 何发













评论
加载更多