新药加速获批的路径都有哪些
一个创新性好、疗效突出、安全性好、社会需要的新药,就像上文中那位天赋异禀的优等生,会享受到很多优待(如下图)。
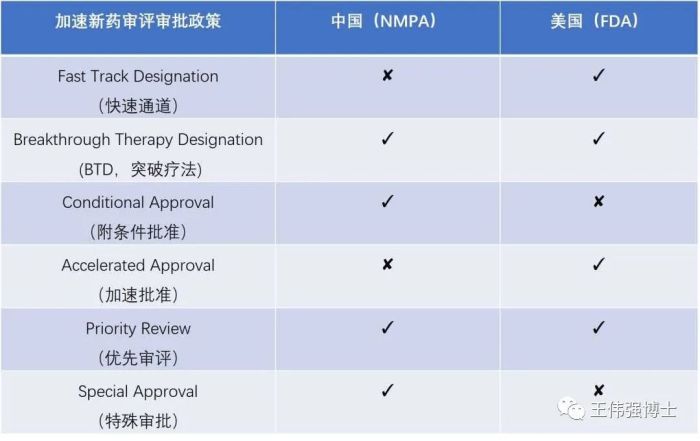
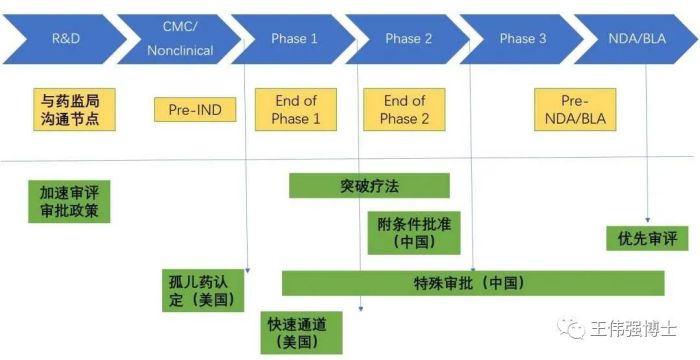
1. 突破疗法(breakthrough designation, BTD)
“突破性疗法”认定,无论是在中国还是美国,都是一种很有利的政策,可以加快新药的研发进程和上市评审。
获得“突破性药物”认证的药物开发,会得到药监局官员更加紧密的指导,确保最快时间给患者提供新的治疗选择。
要申请获得“突破性疗法”认证,必须有足够的临床有效性数据,表明该研究药物比现有疗法有显著优势。
中国药监局和美国药监局都有“突破性疗法”申请政策,获得认定意味着该药物有较高的上市可能性,还会有各种利好政策陆续出台。
2. 附条件批准(conditional approval)
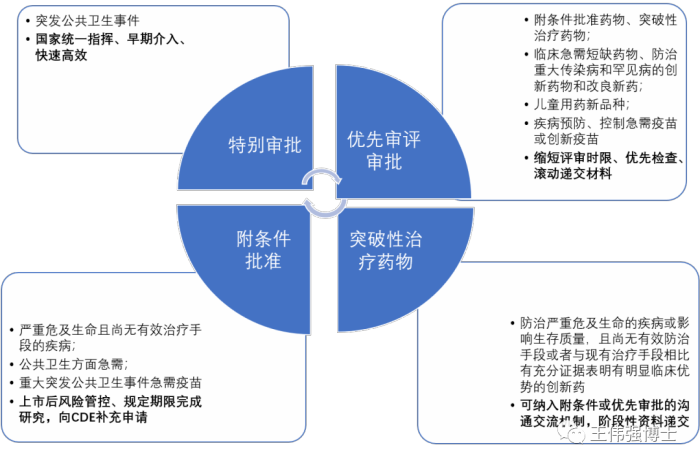
"附条件批准"的政策,是中国NMPA允许针对严重危及生命且尚无有效治疗手段的疾病、公共卫生方面急需的药品,在规定申请人必须履行特定条件的情况下,基于替代终点、中间临床终点或早期临床试验数据而批准上市,以缩短药物临床试验的研发时间。
替代终点可以是实验室检查项目、放射影像学、体征或其他指标,它们本身并不衡量临床获益,但可以预测临床获益。
例如,在某些癌症类型中,肿瘤缩小(如ORR,客观缓解率)的影像学证据有可能预测整体生存率的改善。
美国FDA没有这种"附条件批准"的政策。
3. 优先审评(priority review )
有明显潜在的临床价值的药物,可以申请优先审评程序,该程序可以分为6类:
1)临床急需的药品,比如创新药和改良型新药,用于治疗重大传染病和罕见病等疾病;
2)符合儿童生理特征的儿童用药新品种、剂型和规格;
3)疾病预防、控制急需的疫苗和创新疫苗;
4)纳入突破性治疗药物程序的药品;
5)符合附条件批准的药品;
6)国家药品监督管理局规定的其他优先审评审批的情形。
如果一种药物能够显著改善有效性或安全性,比如用于防治严重疾病,有证据显示比现有治疗方式有明显优势,那么新药研发公司可以在递交新药上市申请(NDA/BLA)或有效性补充申请(efficacy supplement)时,向药监局申请优先审评认定。
中国NMPA会在130个工作日(标准审评时间是200个工作日)内完成上市审批,而美国FDA会在6个月(标准的审评时间是10个月)内完成上市审批。
4. 特别审批(Special approval)
特别审批程序,是中国药监局推出的一项优惠政策,可以让突发公共卫生事件发生或存在威胁时,应急所需药品能够尽快获得批准上市。
美国FDA没有此优惠政策。
药品特别审批程序极大的缩减了药品审批的时限,要求国家药监局应在24小时内决定是否受理申请人的注册申请,并在24小时内组织技术审评和现场核查。
现场核查报告应在5日内完成,首轮技术审评应在受理申请后15日内完成,如要求申请人补充资料,补充资料后最长7日内必须完成技术审评。
技术审评完成后,3日内作出批准临床试验的决定。
临床试验完成后,申请人提交药品上市的申请,国家药监局再次组织现场核查、药品检验、技术审评,时限与临床试验审批相同。
2009年流感病毒裂解疫苗只用了87天就从获得病毒株开始研发至获批上市,可见这项政策的便捷性。
5. 快速通道
美国FDA推出的快速通道(Fast Track)政策旨在加快新药的审评审批过程,而中国药监局暂时并没有类似的优惠政策。
申请快速通道需要提供证据,表明新药对未被满足的临床需求有潜在效果。
这些支持数据可以来自早期临床试验数据,也可以来自非临床研究数据,比如药物在相关疾病模型上取得的非临床研究有效性数据或者能够支持药物机理的非临床研究数据。
有些新药在进入临床试验前或者早期临床试验阶段就凭借非临床研究数据获得快速通道资格。
相比之下,要获得突破性疗法认定(BTD),申报方必须至少在早期临床试验中就取得有效性数据。
6. 孤儿药
事实上,孤儿药认定不是FDA加速新药审评审批过程中的优惠政策,但从客观上来说,它已经帮助了许多新药更快地获得批准上市。
孤儿药认定(Orphan Drug Designation,ODD)是美国FDA孤儿药产品开发办公室授予的一种资格认定,它针对的是影响美国人群少于20万的罕见病。
获得该认定的药物可以享受7年的市场独占权、免除NDA/BLA申请费(PDUFA)、临床研究费用可以享受25%!的(MISSING)税收减免,以及可能免除部分临床数据的申报要求等一系列政策优惠。
虽然中国药监局没有孤儿药认定,但它仍然有针对罕见病药品上市的优惠政策。
不同的是,中国没有明确地界定患有罕见病的患者人数,但制定了罕见病目录。
结语
中国和美国药品监管机构出台的各项优惠政策,为新药审评审批的加速发挥了重要作用。
这些政策极大的缩短了新药的上市时间,使其能够更加快速地进入市场,为病人提供更有效的治疗。
未来,相信药品监管机构还会通过更多的政策和措施,积极创新,完善审评审批制度,加快新药上市,为患者提供更多的治疗选择。
收录于合集#新药研发 7 #医药4
内容来源:王伟强博士
责任编辑:胡静 审核人:何发
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多