除菌滤器完整性测试允许失败几次?
一、法规要求
1.《2010版GMP附录1-无菌药品》第七十五条(三):除菌过滤器使用后,必须采用适当的方法立即对其完整性进行检查并记录。常用的方法有起泡点试验、扩散流试验或压力保持试验。
2、《2020版欧盟GMP附录-无菌产品生产》规定:“灭菌后的过滤器组件的完整性应在使用前通过完整性测试进行验证,以检查由于过滤器使用前的准备造成的完整性破坏或降低。用于进行液体除菌的除菌级滤器应当在使用后进行无损完整性测试,随后再将滤器从滤壳中取出。测试结果应当与过滤器在验证期间建立的微生物截留能力关联起来。使用的测试实验包括起泡点法,扩散流法,水侵入法或压力保持试验。
3、《2004版FDA关于无菌工艺生产的无菌药品的指南》规定:完整性测试可以在过滤工艺前进行,并应当在过滤以后进行日常测试。过滤后 通过完整性测试检测过滤过程中可能出现的泄露或破损是非常重要的。
4、《PDA Technical Report》 TR26《PDA技术报告(26版)》规定:“现行的GMP要求过滤器及过滤系统在使用前及使用后均需要进行完整性测试”。
二、个人理解
1、法规只是说使用后必须要进行完整性检查,并没有说允许测试失败几次。但是,如果测试不通过,就一直不断重复测试,直到测试通过,这点监管部门肯定是不认可的。我见过一个过分的企业,3次测试不通过就换一台完整性检测仪再次测试,还是不通过,就换测试方法,最后测试通过了。最可气的是,前面这么多测试过程都没有记录,只有最后成功通过的打印条。我们是在完整性测试仪的历史数据中找到这些记录的。
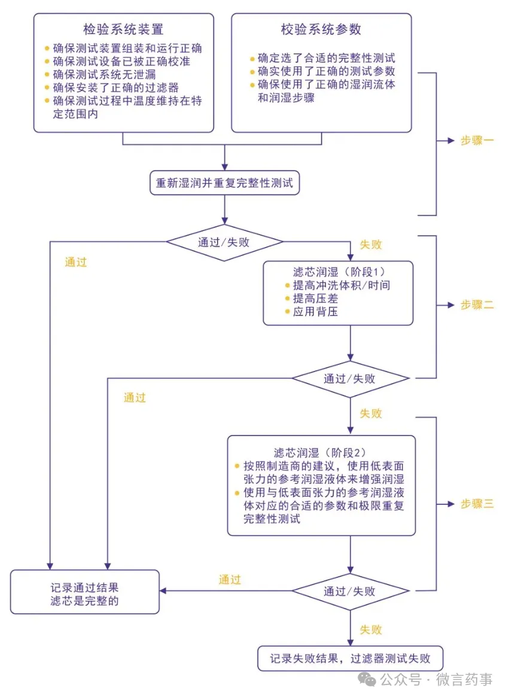
2、所以允许失败几次?《PDA Technical Report》 TR26提供了一个完整性测试失败分析判断树。从判断树种可以看到第四次失败就判定为过滤器测试失败。如果企业没有更好的依据,那最好不要超过四次,不然检查发现这个问题,肯定给落个评估不充分的缺陷。
当然,也有企业说如果滤器是损坏的,那么测多少次都是不通过。所以不限制测试次数。但是企业不能提供依据。我个人也是不认可这点的。
三、看案例
去很多企业检查问企业,如果完整性检测失败了怎么办?企业斩钉截铁地说,那这批肯定就废了,不能放行。再问,这几年测试有失败过吗?有没有记录?企业说,没有失败过。再问,完整性检测仪能保存历史数据吗?有没有看过历史数据?企业就吭哧吭哧不敢回答了。
如果是常态化生产,一年下来没有1次测试不通过,那我就会怀疑数据真实性完整性。作为企业的QA、质量经理、质量负责人,对关键结果都应该有足够的敏感性,至少要抽查一些数据。操作人员有没有把失败的检测结果记录下来?如果没有,是规程没规定、培训没到位、还是故意不记录?
近日某省药监局公布一则行政处罚信息,某药企质量负责人和显微鉴别岗位,分别被罚没十五万六千和三万七千元。原因是部分中药显微图谱擅自套用其他图谱,检验记录不可追溯,这是显微鉴别岗位的问题。而质量负责人对显微岗位的人员没有对应的技能培训,且没有及时发现套用图谱的问题,这是质量负责人的问题。
质量负责人没有及时发现套用图谱,所以被行政处罚。现在“处罚到人”的情况越来越常见。但是,国家认为还不够,越来会有越来越多的“处罚到人”。所以,“质量负责人”们对自己负责吧,认真一些。
微言药事
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多