无菌医疗器械包装质量控制要点(上)
1 无菌医疗器械包装材料分类
目前市面上的无菌医疗器械包装材料,按照材料可分为纸、聚烯烃非织造布、塑料膜、玻璃等;按照包装形式可分为塑料袋、纸塑袋、纸质袋、铝塑袋、吸塑盒等。选择包装,需要考虑很多的因素,如包装系统与产品的适应性、与使用时的适应性等,还应结合有效期、灭菌、运输等综合评价,包括安全性要求、微生物屏障要求、密封完整性要求、密封强度要求、外观要求、印刷要求等。
2 无菌医疗器械国内外标准
我国无菌医疗器械虽然已走过 30多年的历程,早期的无菌医疗器械包装发展相对有些落后。在 2005 年,我国等同采用了 ISO 11607-1997 发布了 GB/T196663-2005《最终灭菌医疗器械的包装》标准,该标准的发布,标志着无菌医疗器械包装标准体系的正式建立,推动了我国无菌医疗器械包装的迅速发展。
2.1 国外常用标准
国外通用标准 ISO 11607 分为两部分,ISO 11607-1 第 1 部分:材料、无菌屏障系统和包装系统的要求 [1] 以及 ISO11607-2 第 2 部分:成型、密封和装配过程的确认要求。该标准是无菌医疗器械包装的一个纲领性标准。此外,还有欧盟发布的 EN 868 系列,ASTM 系列。比较常用的如:ASTM F1980-21 StandardGuide for Accelerated Aging of SterileBarrier Systems and Medical Devices ;ASTM F88/F88M-23 Standard TestMethod for Seal Strength of Flexible BarrierMaterials ;ASTM F1929-2015 StandardTest Method for Detecting Seal Leaks inPorous Medical Packaging by Dye 等。
2.2 国内常用标准
2005 年, 我 国 等 同 采 用 了 ISO11607-1997 并发布了 GB/T 19633-2005《最终灭菌医疗器械的包装》,之后我国又先后转化了 ASTM 等试验方法标准, 形 成 了 YY/T 0681 和 YY/T 0698的系列。后来不断更新标准年份,目前GB/T 19633 已经随着 ISO 11607 更新为两部分,即 GB/T 19633.1-2015 最终灭菌医疗器械包装第 1 部分:材料、无菌屏障系统和包装系统的要求 [2] 以及 GB/T19633.2-2015 最终灭菌医疗器械包装第 2部分:成形、密封和装配过程的确认的要求 [3]。
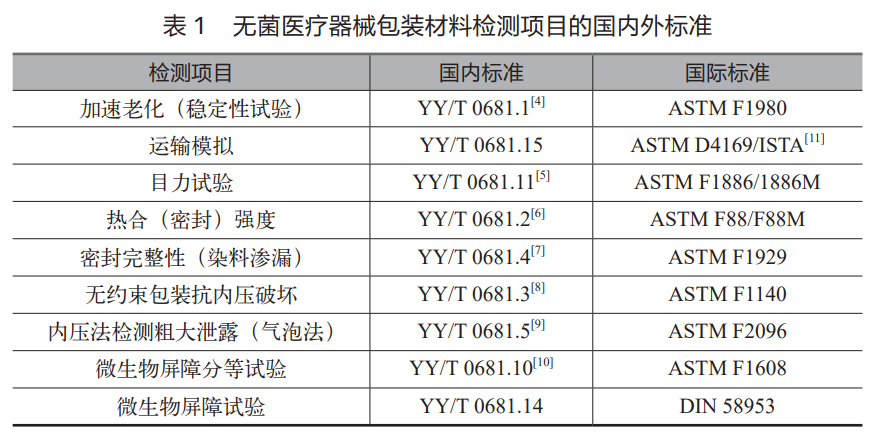
现在行业里比较常用的无菌医疗器械包装材料,比如特卫强、PE/PET 复合膜、吸塑盒,国内外测试参考使用的标准通常见表 1。
3 无菌包装质量控制
无菌包装最终要保护器械的安全和有效,在包装研发设计时,其流程和常见形式包括设计输入阶段,设计输出阶段以及设计验证阶段。
3.1 无菌医疗器械包装设计的输入阶段
在包装研发设计时,设计输入通常来自于 DFEMA、PFEMA、产品的需求、法规标准以及市场同类包装的分析等。DFEMA、PFEMA 通过设计阶段和生产过程阶段,分析包装系统所有组成的失效模式和后果情况。比如一些骨科植入产品,通常产品重量大、有棱角。这时候去选择无菌包装,首先要考虑的无菌包装是吸塑盒类。吸塑盒相对袋类、膜类等包装,保护性能更加卓越,可有效避免医疗器械在运输、存放等环节被破坏,保证了产品的完整性及功能性。但是吸塑盒也有缺点,如材料价格相对较贵,封口设备价格较高。
在产品的需求和法规标准上,选择无菌包装的时候首先要满足法规标准,法规标准是最低限度要求。包括对无菌包装的物理性能、化学性能、生物相容性、毒理学特性、微生物屏障、与成型与密封过程的适应性、与预期灭菌过程的适应性、与标签系统的适应性、灭菌前后储存寿命(加速老化)的评价、运输过程的评价。在产品的需求上,要兼顾考虑产品打开使用前的携带、交叉污染、微粒污染、方便快捷的使用操作等。
对于市场上成熟的同类包装系统,通常也有参考意义。毕竟医疗器械无菌包装主要材料也就是固定的几类,同类产品在市场上经受了考验,其材料特性也可以借鉴。一般器械本身的价值属性也决定着包装材料的选用。我们的医疗器械是低值耗材还是高值耗材,包装形式是否会对现有包装设备适用,相应的检验设备是否适用,灭菌方式是否变化等情况。
3.2 无菌医疗器械包装设计的输出阶段
有了设计输入,就会有输出。接收输入并转换为输出的活动,都是一个过程,设计开发输入阶段的输出,为设计过程开展的一系列活动提供了依据。内容包含设计文件以及工艺的输出。
在设计文件中,首先要输出的是设计方案、图纸、质量标准和印刷文件。在生产工艺上要输出设备及工装、图纸、SOP、工艺流程图、参数等信息。在设计方案里要充分考虑对于无菌包装材料或预成型无菌系统接收准则或技术要求。
3.2.1 设计方案、图纸、质量标准和印刷文件输出
在设计方案里,要明确使用的材料。例如无菌包装是选择吸塑盒还是特卫强袋,或者 PE 复合膜等等。还有为什么选择这些材料,这些材料包装产品的优势是什么。如下是常用的包装方式:
纸纸袋,是纸 / 纸 PP 或纸 / 纸 PE为材料,一面是多孔纸,主要是为灭菌提供气体通道,另一面是不涂层或涂层的复合材料(PP 或 PE),纸面材质为医用透析纸。特点是袋体承重小,价格低廉。
塑料袋,PA/PE/PET 尼龙复合膜和易撕膜 / 复合膜 / 共挤膜的组合,这类材料可视内包装器械产品,适合辐照灭菌。
纸塑袋,特卫强纸 + 复合膜,特卫强纸有涂层和非涂层模式,复合膜有PE/PET,CPP/PET 等,这类材料承载重量较轻,可视内包装器械产品,价格稍高。
铝纸复合袋,材料主要是铜版纸 /AL/PE+ 铜铝纸 /AL/PE,袋体避光、耐腐蚀、阻隔性强。
顶头袋,膜 / 复合膜,头部位置是同包装袋等宽度或较小宽度的透析纸,材料一般是多层共挤 PE 和特卫强纸,或者 PET/PE 和特卫强纸。特点是细菌阻隔能力强,透气性良好,洁净剥离。可用于环氧乙烷和辐照灭菌、等离子灭菌等。有大体积器械考虑使用,相对价格也比较高。
吸塑盒类,由吸塑盒和透析纸组成,盒体材质一般是 PET 或者 PETG,盖材是医用涂胶纸,特卫强纸。这类材料密封性能好,保护性好,便于运输。可用于环氧乙烷和辐照灭菌、等离子灭菌等,但是价格较高。
此外产品和无菌包材的灭菌方式最终也决定了选用哪种合适的无菌包装。如果后期灭菌使用环氧乙烷,那无菌包装就要选择透气材料,如纸、复合透析纸、无纺布、通气型硬质容器等,不能选择非透气的材料,如 PE/PET 复合膜、金属箔、聚氯乙烯等。常用的灭菌方式除了环氧乙烷,还有高温蒸汽灭菌、辐照灭菌、过氧化氢灭菌等。这些灭菌方式的优缺点如下:
环氧乙烷灭菌,环氧乙烷(EthyleneOxide, EO ),分子式 C2H4O,优点是低温低湿,可在常温下杀灭各种微生物,EO 气体的蒸气压比较大,所以对灭菌物品的穿透力强,可以灭菌到包装和器械深处,杀菌范围广,成本低;缺点是灭菌时间长、效率较低,有化学残留,环境也有污染。适用的包装需要是透气包装。影响环氧乙烷灭菌效果的因素众多,因此没有通用的灭菌参数,为了保证灭菌效果,不同的无菌包装应对灭菌过程进行验证。任何一个因素改变时,都需要重新进行验证,因此需要对过程严格控制,与环氧乙烷灭菌过程控制相关的标准有 GB 18279.1-2015《医疗保健产品灭菌环氧乙烷 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制的要求》[12],GB 18279.2-2015《医疗保健产品灭菌 环氧乙烷 第 2 部分:GB 18279.1应用指南》[13] 等。
高温蒸汽灭菌,优点是成本较低,设备简单,灭菌时间短,无环境污染;缺点是有些不耐高温产品不能使用,灭菌有效期也比较短。适用的包装也是需要耐受高温高湿。一些不耐高温、高湿的物品不能采用,如塑料、皮革、毛皮、内镜等,蒸汽无法穿透的物品也不能用,如凡士林、甘油、粉等是湿热隔绝者,蒸汽只能达到表面,不能穿透到内部进行灭菌。
辐照灭菌,优点是灭菌彻底,操作安全,没有残留;缺点是辐照灭菌的建设成本比较高,并且对放射源储存的安全性要求高。适用的包装可以是不透气的包装,如铝箔,复合膜类。辐照灭菌主要是利用 X 射线,Y 射线和电子束杀灭微生物。穿透力较强,可直接用于成品包装。在灭菌过程中温度几乎不变,射线可以接到达器械内部,灭菌后的产品可以直接使用。但钴 60 射线对高分子材料和包装材料有一定的破坏性,对人和环境也存在潜在污染。与辐照灭菌过程控制相关的标准有 GB 18280.1-2015《医疗保健产品灭菌 辐射 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制要求》,GB 18280.2-2015《医疗保健产品灭菌 辐射 第 2 部分:建立灭菌剂量》[14],GB/T 18280.3-2015《医疗保健产品灭菌 辐射 第 3 部分:剂量测量指南》[15],GB 18280-2007《医疗保健产品灭菌-辐射》[16],YY/T 0884-2013《适用于辐射灭菌的医疗保健产品的材料评价》[17] 等。
过氧化氢灭菌,优点是安全、快速,没有残留毒性物质;缺点是成本高昂,穿透力差,灭菌条件严格。适用于特卫强包装。
产品的特性也是选择无菌材料的重要依据,单件小件物品采用纸塑袋包装材料或吸塑盒包装材料,如牙科和骨科小部件等;大件物品或数量多的灭菌包则选择医用无纺布、硬质容器包装材料,如一些器械包,则选择纺织类包装材料。
在设计方案里确定选择的材料和使用的灭菌方式后,结合产品的重量、形状等特性,输出合适的无菌包装的尺寸。设计出适合预期用途的形状,包含内部保护性材料。使用的材料的名称、料号、长宽、厚度、公差范围均清楚的标识于图纸。图纸可收录于质量标准中,用于来料检验控制指标之一。
在质量标准中,需要更加详细的描述包材的物料信息、名称、料号、规格、组分、图纸、储存条件、有效期限、进货检验内容、年度检验内容等,包括印刷要求。
3.2.2 生产工艺设备及工装、图纸、SOP、工艺流程图、参数输出
生产设备以及工装,是基于无菌材料的选择。袋类的材料热封设备,可以采用脉冲加热,或者使用电动或气动压合的热封方式进行热封,热封设备比较小型,也比较通用,可对透析袋、塑料袋、铝箔袋等材质的包装袋进行热封处理。也有些非透气包装袋,结合产品需求,会进行抽真空和充氮气的操作。这类包装操作过程会用到一些合适的模具工装,设备相对只进行封口的设备,结构更复杂,体积也要大很多。
抽真空充氮气封口机的工作原理主要分为以下几个步骤:抽真空、充氮气、封口。首先,抽真空过程利用真空泵将袋内的空气抽取,降低袋内压力,为后续充氮气做好准备。接着,通过充氮气系统,向袋内注入一定量的氮气,达到防氧化的目的。最后,通过封口装置将袋口加热,将塑料薄膜封闭,确保医疗器械包装袋的密封性。
吸塑盒封口设备一般体积较大,采用加热和压力控制技术,实现封口的快速、均匀和牢固,确保产品的密封性,吸塑封口机通过恒温恒压式封口,也能够有效地保障产品的清洁度和安全性。
了解设备和工装的结构以及作用原理,生成工装图纸、模具图纸,根据设备特点生成操作规范。SOP 生效后,遵守相关的安全操作规程,保证操作人员和设备的安全。合理安装、维护设备,确保设备在正常工作状态下运行。
在工艺流程图里,确定工序和工序特点。无菌包装是在洁净室环境下进行,对成品的质量、性能、功能、寿命、可靠性及成本等有直接影响,同时该工序是质量需进行破坏性试验或采取复杂昂贵方法才能测量或只能进行间接监控的工序,通常把无菌包装封口定义为关键工序和特殊工序。对应的工艺规范、检验规范都要匹配在工艺流程图里。
无菌包装封口的参数输出,需要识别关键过程参数并确定各过程参数得特性曲线,如加热温度、冷却温度、压力、封口速度、预热时间等,利用高一级精度设备对以上参数的特性曲线进行确认。调节合适的封口温度、封口压力和封口时间,确保封口牢固、不易破损。
3.3 无菌医疗器械包装设计的验证阶段
设计验证是对设计输出项目进行的检查,确定输出活动满足了规定的要求。设计验证包含了原材料的评价、老化验证、运输模拟验证;工艺上要对设备验收、工艺验证、过程确认、批量的验证、软件确认。
3.3.1 原材料的评价、老化验证、运输模拟验证
根据无菌包装材料的不同,评价项目也不一样,通用的一些项目如下。
微生物屏障能力:评价微生物屏障特性的方法分两类:①适用于非透性材料的方法(ISO5636.5:2003 中规定的葛尔莱 Gurley 法、GB/T 458:2008 肖波尔法)。②适用于透气性材料的方法(YY/T 0681.10 透气包装材料微生物屏障试验、ISO 11607.1 中 5.2.3[1] 试验方法、以 0.45 μm 为参考值进行孔径评价法(一般认为对微生物过滤的最大孔径为 0.45 μm);微生物屏障还有 YY/T0681.14-2018《无菌医疗器械包装试验方法第 14 部分:透气包装材料湿性和干性微生物屏障试验》(等同 DIN 58953-6:2010)。
生物相容性和毒理学特性:生物相容性的要求参考 GB/T 16886.1,一般选择按照表面器械的生物学评价项目来进行包装材料的生物学评价是可以接受的选择。通常比较常规的生物三项,如致敏、皮肤刺激、细胞毒。包装材料在进行生物相容性评价前应经过更严苛的灭菌工艺灭菌。当无菌医疗器械包装中有液体时,还应对包材溶出物质进行评价。
物理性能:如抗张强度、厚度差异、抗撕裂性、透气性和耐破度、微粒污染水平、初始污染菌等。这些物理测试项目可以是供应商提供资料,使用方选择性进行验证评价,有条件的工厂实验室进行收货检验,不具备测试条件的企业,也可以委托有资质的第三方进行测试评价。其中材料的微粒污染水平、初始污染菌是风险项目,应重点检测评价。
化学性能:结合无菌包装材料特性,测试溶出物、pH、氯化物、重金属、易氧化物、硫酸盐含量,以满足包装系统或灭菌过程的要求 [3]。
涂胶材料的要求:涂胶材料的涂层应是连续的;涂胶量要符合标称要求;密封后应满足最小密封强度规格;材料的基本重量(每单位面积质量)应与规定值一致 [18]。
老化验证分为实时老化和加速老化两部分 [4]。
加速老化参考的标准是 ASTMF1980 或 YYT 0681.1。由于产品储存期( 有的产品 8 年 ) 比较长,如果按实际环境储存条件进行检测需要很长的时间才能获得结果,为了在实时有效期结果获得以前,有必要进行加速老化实验提供确定有效期的实验数据。加速老化试验的同时也要进行实时老化 [4]。
加速老化试验的核心理论是:材料退化过程中的化学反应遵循阿列纽斯(阿伦尼乌斯)反应速率函数,均相过程的温度每增加或降低 10℃,其化学反应的速率加倍或减半 [4]。
基于该理论,可通过下列公式估计加速老化因子。式中,AAF 指加速老化因子,TAA 指加速老化温度,TRT 指环境温度。Q10 指老化因子,一般通过在各种温度下对材料进行试验来确定该值。通常,取 Q10 等于 2,是计算加速老化因子的通用和保守方法 [4]。
参考文献
[1] ISO 11607-1 Packaging for terminallysterilized medical devices - Part 1:Requirements for materials, sterilebarrier systems and packaging systems
[2] GB/T 19633.1-2015《最终灭菌医疗器械包装》-第 1 部分:材料、无菌屏障系统和包装系统的要求
[3] GB/T 19633.2-2015《最终灭菌医疗器械包装》-第 2 部分:成形、密封和装配过程的确认的要求
[4] YY/T 0681.1 无菌医疗器械包装试验方法 第 1 部分:加速老化试验指南
[5] YY/T 0681.11 无菌医疗器械包装试验方法 第 11 部分:目力检测医用包装密封完整性
[6] YY/T 0681.2 无菌医疗器械包装试验方法 第 2 部分:软性屏障材料的密封强度
[7] YY/T 0681.4 无菌医疗器械包装试验方法 第 4 部分:染色液穿透法测定透气包装的密封泄漏
[8] YY/T 0681.3 无菌医疗器械包装试验方法 第3 部分 无约束包装抗内压破坏
[9] YY/T 0681.5无菌医疗器械包装试验方法第 5 部分:内压法检测粗大泄漏(气泡法)
[10] YY/T 0681.10无菌医疗器械包装试验方法第 10 部 分:透气包装材料微生物屏障分等试验
[11] ASTM D 4169运输容器和系统的性能试验规范
[12] GB 18279.1-2015《医疗保健产品灭菌环氧乙烷 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制的要求》
[13] GB18279.2-2015《医疗保健产品灭菌 环氧乙烷 第 2 部分:GB 18279.1应用指南》
[14] GB 18280.2-2015《医疗保健产品灭菌 辐射 第 2 部分:建立灭菌剂量》
[15] GB/T 18280.3-2015《医疗保健产品灭菌 辐射 第3部分:剂量测量指南》
[16] GB 18280-2007《医疗保健产品灭菌-辐射》
[17] YY/T 0884-2013《适用于辐射灭菌的医疗保健产品的材料评价》
[18] YY/T 0681.8 无菌医疗器械包装试验方法 第 8 部分:涂胶层重量的测定
撰稿人 | 曹旭
责任编辑 | 邵丽竹
审核人 | 何发
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多