要上市,先变更,CAR-T变更新政解读
随着细胞治疗商业化时代的到来,越来越多的企业步入确证性临床或报产上市阶段。而这一阶段中,工艺变更及可比性研究正在逐渐成为监管侧与企业侧的核心交锋点。国家药监局一直着力完善细胞治疗相关法规与指导原则。此次药审中心(CDE)推出《自体CAR-T细胞治疗产品药学变更研究问题与解答(征求意见稿)》(下文以“《CAR-T变更研究》”代称),正是针对这一问题量身定做的药方。
鉴于此,本文以《CAR-T变更研究》政策为主线,辅以笔者亲历的实战案例,对细胞治疗产品药学变更这一问题进行剖析,以供业内同行参考与交流。
《CAR-T变更研究》政策的推出,除了历史偶然性,更多的是历史必然性。
无论是笔者曾深度参与的全球首款CART产品Kymriah,还是目前我国已经上市的2款和正在报产的4款CART产品,均在IND阶段或上市后发生过重大药学变更,并且都产生了巨大且深远的影响。重大变更发生率为100%,这个数字意味着,对于一家CART企业,我们不需要考虑变更会不会发生,只需要考虑变更何时会发生。换句话说,如果一家CART企业从未碰到变更相关问题,那只能说明这个企业走的还不够远。
变更无法避免是CART本身的新颖性以及行业缺乏经验的共同结果。目前业内比较常见的biotech型企业,无一不是在成立初期依靠少量人员与较为简易的生产车间开展工作,直到获得IND批件或IIT临床数据等成果后,才有意愿和能力去招募专业人才以及扩建产能充足的厂房。CDE的一位部长就曾一针见血的指出:
“开展CAR-T细胞研发的企业,绝大多数是创新型企业。这类申请人从事药品研发时间短,很多企业第1 个产品就是CAR-T 产品。这意味着从事CAR-T研发的企业缺少药品开发研究经验和具有相关经验的人才。”
这种缺乏经验与相关人才的发展模式,本质上就是“摸着石头过河”,难免深一脚浅一脚,甚至踩空了呛口水也不稀奇。这种情况下,重大变更的发生也就在所难免了。
变更发生的最佳时机是早期临床(I期)阶段,而不适宜的时机则是确证性临床(II期)阶段。说白了就是临床开始的越早期,变更所带来的代价越小。
不过,变更这个事情很多时候也不是完全由企业所决定的,而极大程度上取决于外部因素。例如,很多企业在拿到IND批件的那一刻,就会因为CDE的要求(一般称为“留作业”)而开展变更。例如将质粒的抗性基因从β-内酰胺换成卡那霉素就是很常见的一个变更。如果临床开展不顺利,例如注册临床疗效不如IIT临床(这在CART类产品中非常常见,包括最基础的二代CD19 CART),企业也往往会首先考虑从工艺角度提高产品质量,这又带来了变更。
虽说确证性临床(II期)不适宜发生变更,但有时也避免不了。例如,全球第一款CART产品Kymriah就在确证性临床阶段发生过重大变更。宾夕法尼亚大学开展Kymriah的II期临床B2205J时,同阶段还在发生宾大向诺华的技术转移,其中同时包含了场地变更和工艺变更。宾大与诺华虽然都知道此时不宜开展如此重大的变更,但事情已经走到这一步了,也只有硬着头皮上。《CAR-T变更研究》政策中提到,
“原则上,应在确证性临床试验阶段开始前完成重大药学变更”
。这里的“原则上”三个字就用的非常精准。
不过,变更发生的越晚,付出的代价也就越大。宾大开展的II期临床B2205J最终仅被FDA认可为次要临床(secondary study),最终Kymriah上市时的关键临床(pivotal study)是之后诺华另做的二期B2202(ELIANA)。如果B2205J可以被认可,Kymriah的上市时间还能更早。这就是因为变更而付出的代价。


|
图1. 宾夕法尼亚大学为Kymriah开展的II期临床B2205J(内部编号14BT022) |
《CAR-T变更研究》政策中明确的提出了变更流程,具体可以分为三步:①制定变更计划;②评估变更事项;③实施可比性研究。最终的目的是要证明“
变更不对产品产生任何不良影响
”。
具体执行变更时,最常采用的策略是头对头对比和历史数据对比。头对头对比主要是单采血等分配对研究,也就是将一份单采血分为两半,分别采用变更前、后的工艺进行生产,或在新、旧厂房中进行生产。单采血等分配对的主要目的是消除供者个体差异(例如年龄、性别、健康状况等)对CART生产带来的影响,因此是《CAR-T变更研究》政策中最为推荐的研究方法,并且应尽量采用患者细胞进行研究。
历史数据对比主要是作为头对头对比的补充,或者在头对头对比不具有可行性时采用。
《CAR-T变更研究》政策中花了大段篇幅来描述变更计划,并再三强调企业应“
提早制定CAR-T产品变更研究的研发计划和策略,减少非预期变更对产品研发进程的影响
”。不过,对于CART类产品的变更计划,企业应辩证的看待。
一方面,CART类产品是很难进行有计划的变更的。如前所述,CART本身具有很大的创新性,而CART行业又缺乏经验。在这种情况下,想要准确预测可能发生的变更并提前制定计划,本身就不太容易。以外,还有一个很关键的问题,就是CART的关键质量属性(CQA)的复杂性(下文专门讨论)。
另一方面,CART产品变更难以预计的情况CDE是了解的,但依然要求企业去做变更计划。这是因为变更计划是应该做的事情,不能因为难做就不去做。或者说,正是因为变更难以预计,所以才更应该认真仔细的做变更计划。譬如预算,尽管知道有些花销是无法预计的,但还是要做预算。如果真的出现计划外的大笔花销,到时候再根据情况申请追加预算即可。变更计划也是一样的道理。
《CAR-T变更研究》政策中提到的
变更评估主要遵循两个原则:1)变更程度;2)距离最终产品的远近。
变更程度越大、距离终产品越近,则变更越重大,企业要做的工作和付出的代价越高昂。
例如,同样是磁珠变更,从Dynabeads微米磁珠换成美天旎的纳米磁珠,其变更程度就大于把Dynabeads换成国产微米磁珠。终产品距离方面,细胞冻存方法的变更就大于单采方法的变更(单采血距离终产品更远),CART培养基的变更就大于慢病毒293T培养基的变更(慢病毒距离终产品更远)。至于质粒工艺相关变更,由于距离终产品非常远,所以一般很难被认为是重大变更。但也有特例。《CAR-T变更研究》政策中提及的唯一一个终极重大变更(CAR序列改变)就发生在质粒环节,也就是“
如果CAR元件的氨基酸序列改变,建议按照新产品进行研究与申报
”。其实,这种情况已经不能称为变更,而是另一个产品了。
《CAR-T变更研究》政策中提到的
可比性研究分为两类,1)自身研究;2)连带研究
。
自身研究主要是局限于变更本身的,比如某个工艺步骤或某种原材料。例如,如果单采程序发生变更(比如医院引进了更新款的单采机),自身可比性研究主要关注的就是单采这个过程,具体包括单采耗时、白细胞收集量等。只要新款单采机能做到多快好省,那变更一般都能通过。
但是,由于CART类产品的新颖性,很多工艺步骤之间存在着千丝万缕的联系,并且有些联系可能还未得到阐明。例如,新款单采机可能会引起单采血内部某些细胞亚群比例的变化,这种变化可能会影响后续的操作(例如血小板含量会影响后续步骤Ficoll离心的效率)。这时候,只局限于单采的可比性研究就不能全面反映问题,而需要连带的对相关步骤、甚至是整个CART生产流程以及质量属性进行可比性研究。
连带可比性研究在CART变更中非常普遍。《CAR-T变更研究》政策就提到了一个很好的例子。对于病毒工艺变更,如果“可能影响到载体的纯度、杂质和生物学活性时,就要开展CAR-T细胞可比性研究”,这就是慢病毒连带到CART的情况。
这种连带的最终极情况,就是工艺变更连带到额外的非临床或临床研究。这种情况往往是由于CART的CQA缺失导致药学研究难以反映临床情况所引发的。例如,在Kymriah的IND期间,就多次因为工艺变更(例如细胞培养添加的白介素从IL-2换为IL-7/IL-15、增加CD25阴选等新工艺步骤)在当时的研究水平下实在是难以评估其影响,而被FDA要求进行额外的临床研究。《CAR-T变更研究》政策中也明确指出,“
现阶段CAR-T产品的质量属性与安全性及有效性之间的关系仍未充分建立,如果变更风险较高……可能需要进行非临床和/或临床桥接研究
”,其监管思路与FDA可谓异曲同工。而这种额外的临床研究一旦出现,企业所付出的代价就会非常大,所以应该极力予以避免。
《CAR-T变更研究》政策开篇就指出,“
生产场地新增/转移、基因修饰系统工艺变更、生产用原材料更新换代以及分析方法的优化
”是最常见的四大变更事项,更是对多种可能出现的变更情况进行了举例。在此整理并列表如下(实际案例一项为笔者添加):
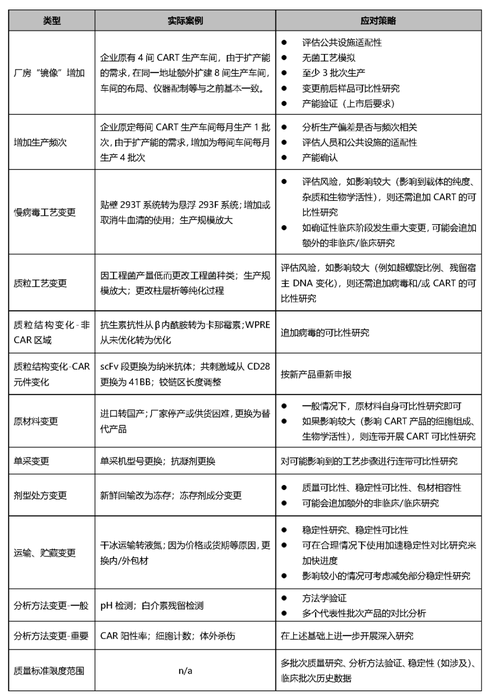
关键质量属性(CQA)对变更后的可比性研究至关重要。原则上说,CQA是可比性研究最主要的验收标准。因为判断多批产品是否可比,主要就是看各批次的CQA是否基本一致。但是,这个情况在CART类产品中往往并不适用。
CART类产品很难确定CQA,尤其是有效性相关的CQA。这一论断的最好例证,就是无论是FDA还是CDE,至今都没有在CQA方面给出任何确定性结论。CQA难以确定的原因比较复杂,但结果却很清楚,就是现有的CQA加在一起也难以反映临床疗效,即使是Kymriah这种已经上市多年的产品(图2)。换句话说,CART类产品的药学和临床评估是脱钩的。生产上判断为高质量的优质批次,在临床上可能根本就无效。
CDE对CQA的问题是完全了解的。《CAR-T变更研究》政策中多处提到,“
现阶段CAR-T产品的质量属性与安全性及有效性之间的关系仍未充分建立
”、“
目前CAR-T产品的关键质量属性仍在不断完善中,其与体内安全性和有效性之间的关系尚需积累更多研究数据
”。此外,还以模糊和隐晦的方式提及,CQA以外还存在着某些重要质量参数,并用“扩展的质量属性”、“其他质量属性”等词语来指代。

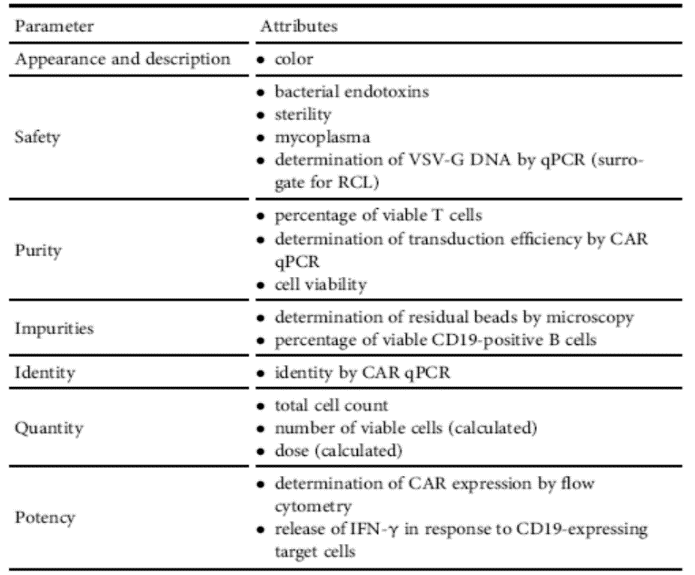
|
图2. 全球首个上市的CART产品(诺华的Kymriah)的CQA列表 |
第一次是在变更时机的章节,
“
原则上,应在确证性临床试验阶段开始前完成重大药学变更
”。这一情况之前已有提及,在此不再赘述。
第二次是在质量标准限度变更的章节,
“
原则上,质量标准变更不应导致产品质量控制水平的降低
”。这里的“原则上”是有缘故的,其根源还是CART产品CQA不明确,因此部分质量标准(尤其是有效性相关项目)有可能存在质量标准制定偏严苛的情况。白血病小女孩Emily Whitehead的主治医生Stephan Grupp博士就多次公开呼吁应放宽CAR-T类产品的质量限度,并在顶级学术期刊上撰文要求调低Kymriah的某些放行标准(图3)。最主要的是,当Grupp博士通过individualized IND、同情性用药等监管途径合理合法的让自己的患者用上了“不合格产品”后,大部分患者依然获得了很好的临床疗效。这其实就说明了这些“不合格产品”其实是合格的,只是质量标准过于严苛了。
不过,这条规则很容易被滥用,因此监管机构对其会非常慎重。因此,除非有非常确凿的依据,否则企业不应过度考虑如何放宽质量标准,尤其是安全性相关的质量标准。



|
图3. 白血病小女孩Emily Whitehead的主治医生Stephan Grupp博士发表文章,呼吁放宽CAR-T的细胞活率相关放行标准 |
《CAR-T变更研究》政策的推出,意味着CART相关的政策法规建设进程,正在趋于完善。
我国监管部门对CART类产品的监管历史,主要可以分为3个阶段。在最初阶段,对于CART这个药品“异类”,相关信息晦暗不明,可借鉴的经验也少,监管力度是比较弱的,很多情况往往是企业侧和监管侧商量着来。随着行业发展,监管理念逐渐形成,综合性法规开始出现,其中的代表就是2017年颁布的《细胞治疗产品研究与评价技术指导原则》(试行)。之后,随着“中国药品监管科学行动计划”的启动,监管日趋完善,法规走向细化,从综合性法规延伸到药学、非临床、临床各自的法规,甚至进一步延伸到某一领域的某个具体问题的针对性法规。《CAR-T变更研究》就是在这一时代背景下应运而生的。
变更研究这个话题,重要吗?当然重要。真的很重要吗?其实也未必。
变更研究再重要,也不过是药学研究中的一个分支,甚至都不能算是最重要的分支。而当我国的法规已经延伸到一个领域的各个分支,那无疑意味着对于这个领域的法规体系建设已经趋于完善,整个行业的监管工作正在走向成熟。2016年前CART领域“无法可依”的问题,也将迎来最终的解决之道。
细胞治疗类产品的出现对监管和被监管方同时提出了挑战。目前,我国监管机构已经充分认识到了细胞治疗产品的特殊性,并针对性的制定了多部政策或指导原则,以更好的适应行业需求、促进行业发展。对业内公司而言,对政策法规的系统性解读可以快速捋清监管要求,为自家产品制定清晰的开发计划,并在实际工作过程中做出正确的决定。
特约撰稿人 | 张长风 博士
来源 | 蒲公英Ouryao
编辑 | Jamie
责任编辑:胡静 审核人:何发













评论
加载更多