组蛋白的乙酰化主要发生在赖氨酸残基上,赖氨酸侧链含有氨基,在生理条件下带正电荷,从而能够和含有磷酸基团的DNA紧密结合,被乙酰化后使得正电荷被中和,无法和DNA紧密结合使得染色质结构松散,促进基因的表达。组蛋白的乙酰化动态平衡由大量不同的组蛋白去乙酰转移酶(HDAC)和组蛋白乙酰化酶(HAT)调节。
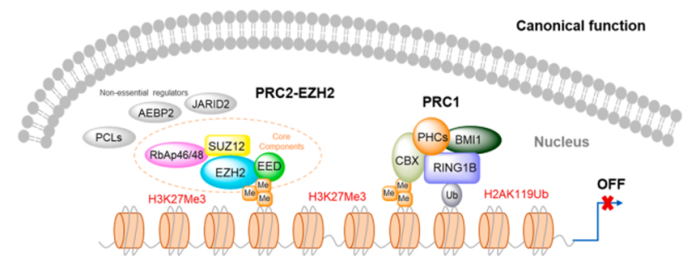
Polycomb 群蛋白(PcG)是一组重要的表观遗传学调节因子,在细胞增殖,干细胞多能性和分化,基因表达异常恶性转化等中起重要作用。PcGs主要含有两种配合物,即多梳抑制复合物1(PRC1)和多梳抑制复合物2(PRC2)。PRC1,由PHCs、BMI1、CBX和RING1B亚基组成,催化组蛋白H2A的单泛素化(图1)1。
PRC2复合物包括EZH2,EED,SUZ12和RbAp46/48(图1)。EZH2是一种组蛋白乙酰化酶,它是PRC2复合物中的催化亚基,可以催化组蛋白H3赖氨酸27(H3K27)三甲基化,诱导染色体凝集,进而抑制靶基因的转录1。
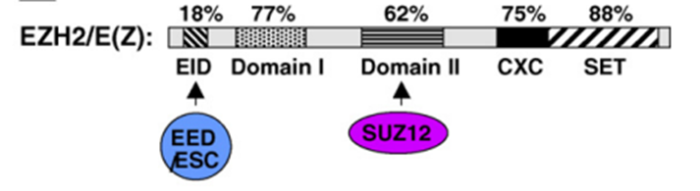
EZH2蛋白质结构主要包含五个结合结构域:EED相互作用域(EID),与PHF1和PCL(结构域I)的结合结构域,与SUZ12结合结构域,富含半胱氨酸的结构域(CXC)和
SET结构域(图2)2
。
近年来,EZH2的病理调控机制见于许多疾病,目前已经开发了许多小分子EZH2抑制剂,包括一些被FDA批准上市的药物和一些已进入临床试验不同阶段的抑制剂。EZH2抑制剂目前主要适应症是非霍奇金淋巴瘤(Non-Hodgkinlymphoma,NHL),而NHL根据细胞形态和病理类型分为弥散性大B细胞淋巴瘤(DLBCL)、滤泡性淋巴瘤(FL)、套细胞淋巴瘤(MCL)和慢性淋巴细胞白血病/小淋巴细胞瘤(CLL/SLL)等亚类。据2018年IARC统计,全球NHL新发病例近51万例,占据所有新发癌症病例的2.8%,发病率呈逐渐上升趋势。
单一从非霍奇金淋巴瘤市场看,到2023年,NHL市场预计将达到将近100亿美元。而小分子靶向药物研发成本相对较低,药物可及性强,一旦EZH2抑制剂在发病率较高的淋巴瘤(比如DLBCL、FL)上取得显著疗效和实体瘤上取得突破进展,将会强势竞争和渗透生物药和其他靶点抑制剂的市场份额。临床需求决定研发方向,EZH2抑制剂前景无限。
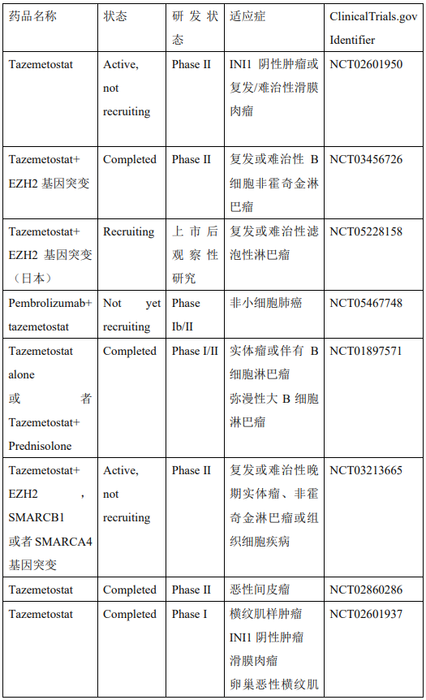
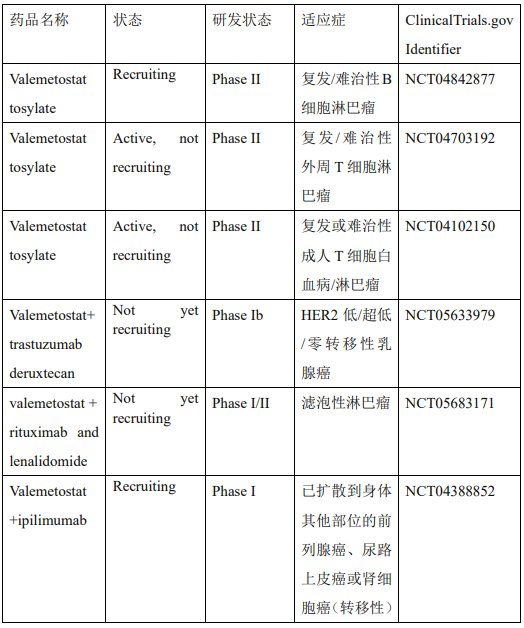
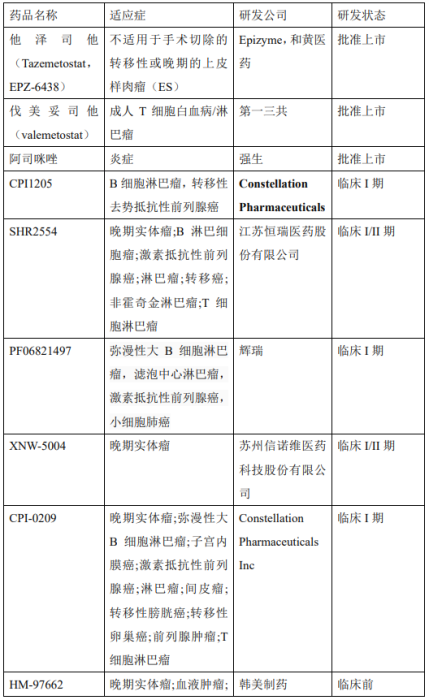
据不完全统计,目前该领域的在研药物四十余种,包括EZH2抑制剂和蛋白水解靶向嵌合体(PROTACs)。其中有3款EZH2抑制剂已获批,分别是和黄与Epizyme共同开发的他泽司他(Tazemetostat,EPZ-6438),第一三共的伐美妥司他(valemetostat),以及强生开发的阿司咪唑,除此之外还有多款处于临床前和临床研究的抑制剂(表1)。
他泽司他(Tazemetostat,EPZ-6438)
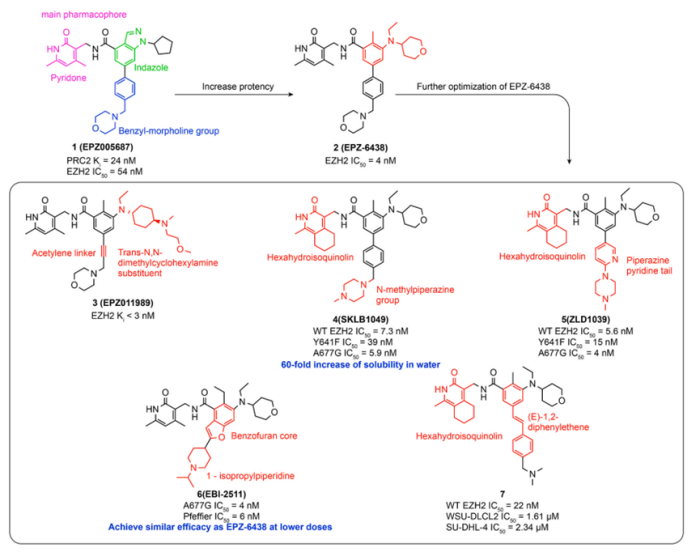
2012年,Epizyme通过筛选175,000的高通量化合物库开发了第一个EZH2特异性抑制剂EPZ005687(化合物1,图3),它的结构通过酰胺键连接吡啶酮和吲唑,Ki 值为 24 nM,IC50值为54 nM3。
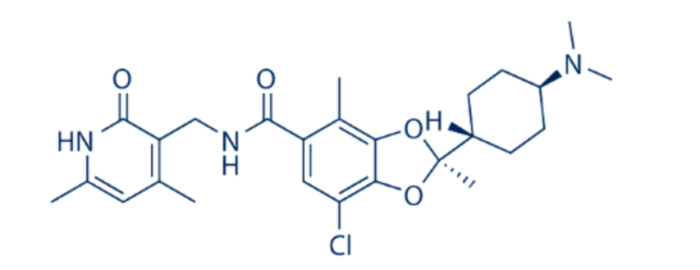
2013年,Epizyme优化了EPZ005687的结构。吡唑的五元环结构断开用苯胺替代,苯胺上的取代基经过SAR分析,发现通过取代乙基和THP获得化合物EPZ-6438(也称为Tazemetostat,化合物2,图3)显示出更强的效力,IC50值为4 nM。与EPZ005687相比,该化合物还显示出良好的药代动力学特性和口服生物利用度。EPZ-6438 对EZH1具有35倍的选择性以及与其他14种 HMT相比,超过> 4500倍的选择性。它对野生型EZH2和EZH2 Y641F、C、H、N、S 和 A677G 突变体表现出很高的体外效力。
后续人们基于EPZ-6438结构进行很多的优化和修饰。2015年,Epizyme报道了一种有效且选择性的EZH2抑制剂EPZ011989(化合物3,图3)。对于EPZ011989,EPZ-6438的四氢吡喃部分转化为4-氨基环己胺,吗啉甲基苯基被吗啉丙炔取代。化学结构的这些变化改善了药代动力学和药效学性质。
2015年,Zhang等报道了一种新的EZH2抑制剂SKLB1049(化合物4,图3),其核心结构为六氢异喹啉。SKLB1049对EZH2野生型、Y641F和A677G突变体非常有效,IC50值分别为7.3 nM、39 nM和5.9 nM。SKLB1049还显示出对EZH1的12倍选择性。
2016年,Song等人报告了一种有效的、高度选择性的、口服生物可利用的EZH2抑制剂ZLD1039(化合物5,图3)。以EPZ-6438结构为基础,用哌嗪吡啶尾取代甲基苯基吗啉,环合吡啶酮C4和C5形成缩合环己烯。
2018年,为了降低EPZ-6438的分子量和复杂性,江苏恒瑞公司的研究人员去除了EPZ-6438左侧的苯基侧链,合成了具有苯并呋喃结构的化合物EBI-2511(化合物6,图3)。它不仅表现出高酶和细胞抑制活性,IC50值分别为4.0 nM和6.0 nM,但也具有良好的药代动力学特性。
2020年,He等人对SKLB1049的哌嗪甲基苯基(溶剂暴露区)进行了结构优化,用于设计以及合成一系列(E)-2-二苯基乙烯衍生物。最初,在SKLB1049的两个苯环之间加入反式乙烯基。进一步探讨了苯侧链上不同取代基的SAR。初步优化发现化合物7(图3)具有有效的酶和细胞抑制活性,(EZH2 WT,IC50 = 22.0 nM;WSU-DLCL2细胞系,IC50 = 1.61 μM;SU-DHL-4细胞系,IC50 = 2.34 μM)4。
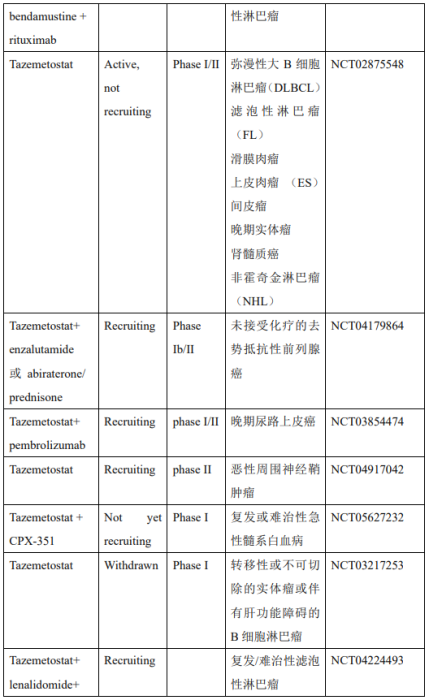
美国时间2020年1月23日,FDA宣布批准全球首个EZH2抑制剂Tazemetostat重磅上市,批准的适应症为不适用于手术切除的转移性或晚期的上皮样肉瘤(ES)。除此之外,目前关于EZH2抑制剂Tazemetostat的临床研究有很多,针对不同的疾病或者与其它药物联合使用(表2)。


伐美妥司他(Valemetostat,图4)是一款新型、高选择性first-in-class的EZH1/2双重抑制剂,2021年获得美国FDA的罕见病用药资格认定,是在全球范围内第1个获得监管批准的治疗成人T细胞白血病/淋巴瘤 (Adult T-cell leukemia/lymphoma,ATL) 的EZH1和 EZH2双重抑制剂。表3显示Valemetostat目前临床研究试验。
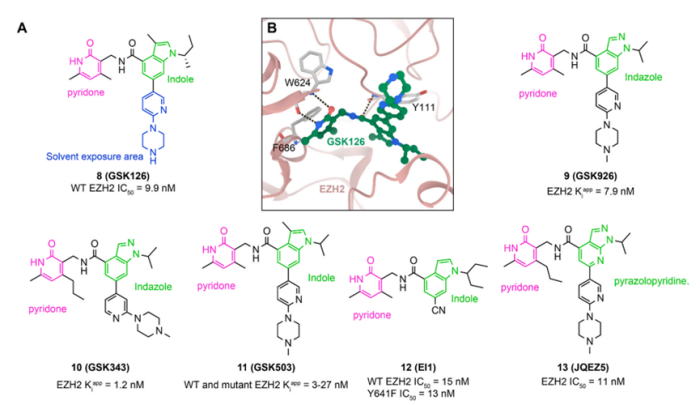
2012年,GSK通过高通量筛选和结构优化发现SAM竞争EZH2抑制剂GSK126(化合物8,图5A)5。GSK126含有与EPZ005687相同的吡啶酮基团,但其吲哚基团与EPZ005687的吲唑基团不同。GSK126对野生型和突变型EZH2均具有有效的抑制活性,Kiapp = 0.5-3 nM。GSK126对EZH2的选择性是其他20种人甲基转移酶的1000倍。其对EZH1的选择性超过150倍。
然而,GSK126缺乏口服生物利用度,因此在小鼠模型和随后的临床试验中需要静脉内(IV)给药。GSK126和PRC2共结晶结构(PDB:5WG6,图5B)显示GSK126中的吡啶酮结构也可以与W624形成两个的氢键。GSK126与F686的苯环有很强的π- π相互作用。其酰胺基团也可与Y111形成氢键。由吡啶基哌嗪组成的“尾部”区域向外指向EZH2和EED之间的溶剂暴露侧。
在SAR的指导下,分别合成化合物GSK926(化合物9,图5A),GSK343(化合物10,图5A)和GSK503(化合物11,图5A)。
同样,在2012年,诺华公司报告了一种有效且选择性的小型分子抑制剂EI1(化合物12,图5A)。El1与抑制剂GSK126具有相同的吡啶酮和吲哚核心结构。
2016年,Zhang等人开发了一种新型有效的EZH2抑制剂JQEZ5(化合物13,图5A)。其核心结构为吡啶酮和吡唑并吡啶。该化合物显示出优异的酶效力
EZH2(IC50 = 11 nM)。
在75 mg/kg下,JQEZ5显示出GSK-126在150 mg/kg时相同的抗肿瘤活性。
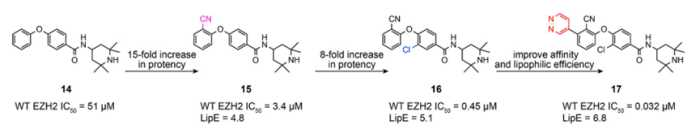
2013年,Constellation Pharmaceuticals公司报告了一系列独特的含有四甲基哌啶基苯甲酰胺骨架的EZH2抑制剂,通过高通量筛选发现苗头化合物14(图6),其对WT EZH2具有51 μM的抑制活性,经过优化发现活性化合物17(图6)。
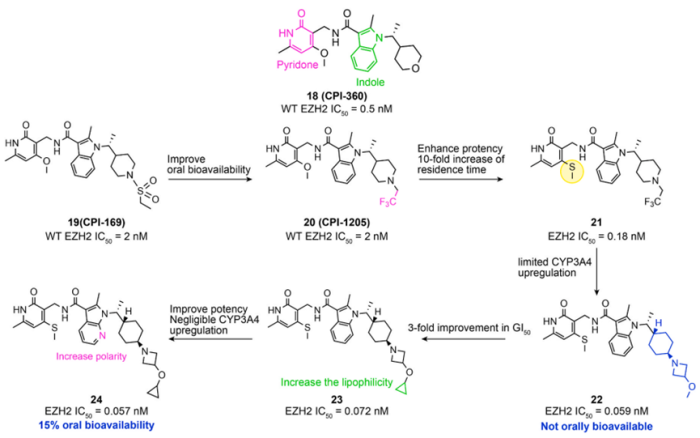
2014年,Constellation Pharmaceuticals公司报道了另一种EZH2抑制剂CPI-360(化合物18,图7)。它是一种酰胺连接吡啶酮和吲哚的化合物。它不仅具有出色的抑制活性(WT EZH2 IC50 = 0.5nM),也显示出显着的药代动力学特征。该公司基于此进行进一步的结构优化发现推进临床研究的化合物CPI-1205(图7)和CPI-0209。表3显示 CPI-1205和CPI-0209的临床在研进展。
表4. CPI-1205和CPI-0209的临床在研进展
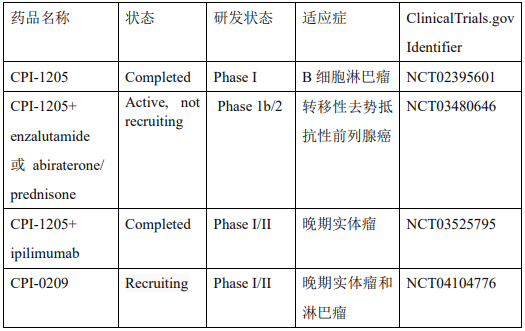
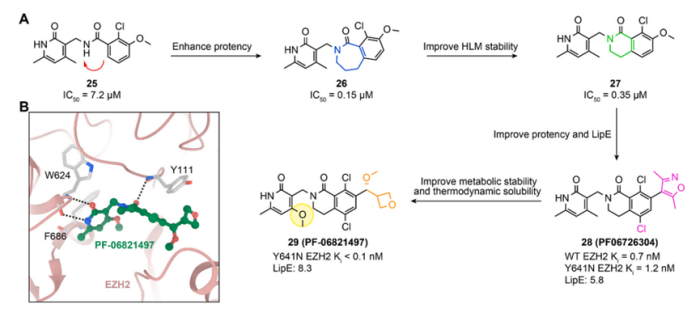
对于一些报道的 EZH2 抑制剂(如 GSK-126、EI1 和 CPI169),吡啶酮通常通过酰胺与双环芳族部分相连。然而,这种结构包含相对大量的平面芳环,通常会损害所获得化合物的溶解度。
EPZ-6438只有一个苯环该中心,它在临床上具有良好的应用前景。基于化合物25,辉瑞的研究团队尝试将苯环与酰胺连接起来,分别得到含有七元内酰胺结构的化合物26和含有六元内酰胺结构的化合物27(图8)。进一步优化得到化合物PF-06821497,目前进入临床I期研究,研究对复发/难治性 SCLC、去势抵抗性前列腺癌和滤泡性淋巴瘤治疗效果(NCT03460977)。
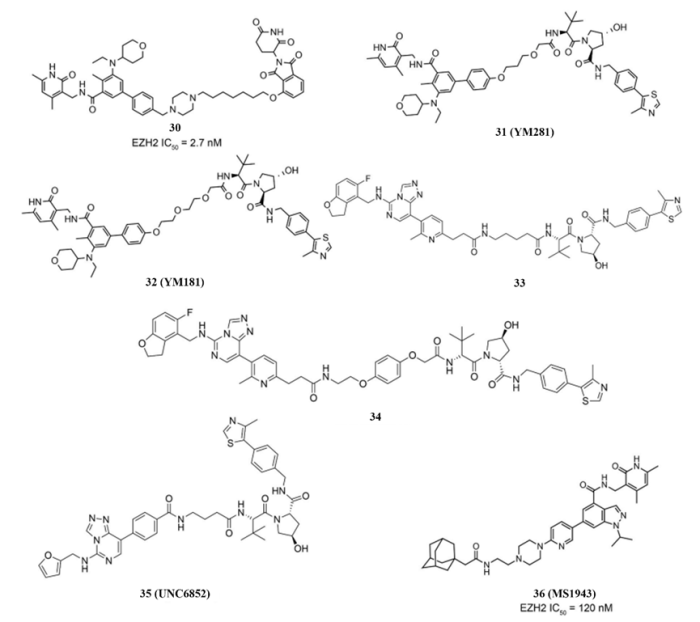
除了EZH2抑制剂外,人们也开发了EZH2的蛋白靶向嵌合体(PROTACs),如图9所示,使用不同的连接链连接EZH2抑制剂和E3连接酶配体(如VHL配体、CRBN配体和金刚烷)。
图9 . EZH2的PROTACs 30-36的结构
作为一种重要的组蛋白甲基转移酶,EZH2介导的表观遗传机制紊乱在癌症的发生和发展中得到了深入研究,证实了EZH2是抗癌靶点。通过对EZH2蛋白结构的表征,揭示其SET结构域是其甲基转移酶的主要部分活性和SAM的结合位点。
随着药物化学和结构生物学的发展,越来越多的靶向EZH2的特异性抑制剂被发现。已经有三款抑制剂被批准上市,而且目前临床在研的抑制剂越来越多。Epizyme的EZH2抑制剂Tazemetostat在不到半年的时间内两次获得FDA批准,是表观遗传癌症药物的里程碑,表明EZH2抑制剂在癌症治疗领域的潜力。
虽然有证据表明单独使用EZH2抑制剂可能对某些肿瘤不是很有效,但替代策略的开发,如联合治疗,多靶点药物设计和开发,EZH2的靶向降解和蛋白质-蛋白质相互作用的破坏是高度期待的。
内容来源:药渡 责任编辑:胡静 审核人:何发













评论
加载更多