有效载荷(payload)的多样性:ADC药物未来开发的关键一环
ADC领域取得的巨大进步主要得益于为特定靶标量身定做的设计。这要归功于几项成就,包括:(1)探索和验证越来越多的靶标;(2)筛选专用于ADC的抗体,重点是交叉反应,利用pH变化有助于与肿瘤的优先结合,促进内吞和FcRn循环;(3)偶联技术的改进,药物与抗体的比更高;(4)有效载荷的多样化(图1)。
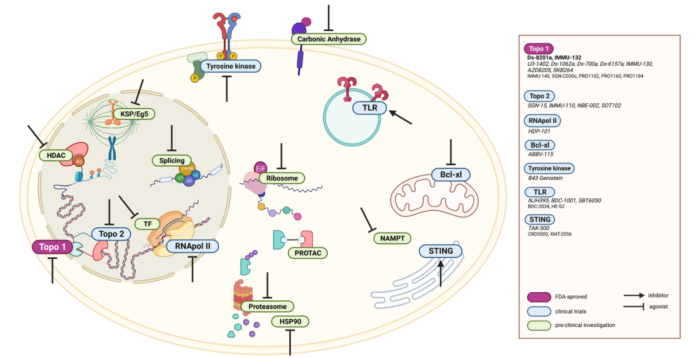
图1. 微管和DNA烷化剂以外的ADC有效载荷示意图
多年来,微管抑制剂和DNA烷化剂一直占据ADC 有效载荷的主导地位。这些有效载荷已使得8个ADC药物获得批准。然而,最近两种基于拓扑异构酶1抑制剂的ADC药物--trastuzumab deruxtecan (Enhertu®)和saituzumab govitecan(Trodelvy®)的批准和临床成功显示了具有不同的作用机制的非传统有效载荷的潜力。这得益于ADC结构的重大突破,通过改进的连接子设计以允许更高的DAR值、更稳定的有效载荷附着和增强的旁观者杀伤活性。在ADC领域的未来发展中,有效载荷多样化有望发挥关键作用,越来越多的处于临床前和临床阶段非传统有效载荷偶联的ADC就是例证。本文就具有不同作用机制的新开发的、有效的、进入临床阶段的非传统有效载荷做一个简要介绍。
成功的有效载荷家族:拓扑异构酶1抑制剂
拓扑异构酶1抑制剂是FDA批准的最新的抗体-药物结合有效载荷家族,最先批准的是trastuzumab deruxtecan,紧接着是sacituzumab govitecan 。
拓扑异构酶位于细胞核内。它们的作用是控制和修复在DNA打开、上游转录和复制过程中发生的DNA超螺旋和缠绕。拓扑异构酶分为两个家族:拓扑异构酶I裂解单链DNA,拓扑异构酶II裂解双链DNA。拓扑异构酶抑制剂特异性地结合到DNA-拓扑异构酶复合体的界面上,从而抑制拓扑异构酶修复机制,导致DNA损伤,从而导致细胞凋亡。然而,最有效的拓扑异构酶抑制剂的效力比微管抑制剂maytansines或DNA烷化剂calicheamicin低100到1000倍,这解释了为什么在最初的ADC设计中对这种有效载荷类别缺乏兴趣。
该有效载荷类别包括基于喜树碱和非喜树碱的化合物。喜树碱(CPT)是一种由五个化学环组成的天然植物生物碱,不易溶于水。几种具有更好生物利用度的衍生物已获得监管当局的批准,如topotecan, irinotecan 和 belotecan。这些药物已经被批准用于几种适应症,包括卵巢癌、肺癌、宫颈癌和结肠癌。Irinotecan的脂质体制剂也已被批准用于治疗晚期胰腺癌。其他几种CPT衍生物也已被合成,例如gimatecan,目前正处于治疗卵巢癌、输卵管癌或腹膜癌的II期评估(NCT04846842)。基于CPT的分子最显著的严重不良事件包括严重水样腹泻、中性粒细胞减少和血小板减少。
CPT衍生物最近被用作ADC有效载荷,因为它们具有中等的细胞毒性效力,IC50值在纳摩尔范围内。它们的效力介于非常有效的抗微管/DNA靶向药物(皮摩尔IC50)和传统化疗药物(微摩尔IC50)之间,后者最初用于最初的ADC,但因缺乏疗效而失败(甲氨蝶呤和阿霉素)。到目前为止,已有两个CPT衍生物成功地与抗体偶联并获得批准:DXd和irinotecan的活性代谢物SN-38。
Exatecan及其衍生物
与CPT相比,exatecan具有更高的活性和更好的溶解性,并且不是ABCC2或ABCG1底物。未偶联的exatecan在几个临床试验中进行了评估,但其糟糕的治疗窗口,具有剂量限制性的中性粒细胞减少和血小板减少以及强烈的胃肠道毒性限制了其应用。
在初步尝试中,exatecan与抗体的生物偶联取得了部分成功,但偶联物显著聚集。Daiichi Sankyo的科学家通过使用一种名为DXd的exatecan的略微修改的乙醇酸衍生物解决了这个问题。结果发现,这种新化合物保留了exatecan的效力,同时使每个抗体成功地偶联了多达8个DXd分子,而且没有明显的聚集。这种deruxtecan 药物-连接子已被用于多个的ADC药物(图2),例如DAR8的DS-8201a(Enhertu®)、U3-1402和DS-6157a,以及在DAR的DS-1062a和DS-7300a,以限制它们的毒性。这些ADC药物中,DS-8201a因为卓越疗效已得到FDA的批准上市,其他的还正在进行临床评估。虽然DXd有效载荷表现出比exatecan甲磺酸盐更低的被动膜渗透性,但它的骨髓毒性较小,因此也因其改善的安全性而被选中。
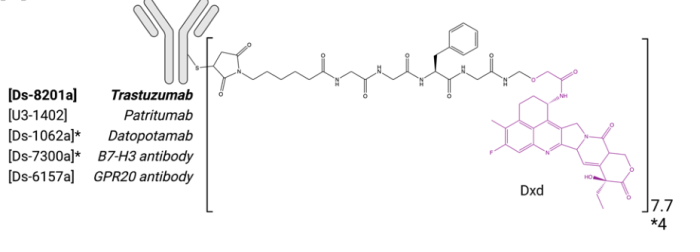
图2. 基于deruxtecan 药物-连接子的ADC药物
最近,exatecan已被作为潜在的ADC有效载荷进行临床前探索,这要归功于亲水性可切割连接子的开发,这些结构能够绕过化合物的疏水和聚集特性。这使得exatecan可以在较高的DAR值下偶联,而不会干扰ADC的药代动力学特性。这些ADC在肿瘤移植瘤中表现出很强的抗肿瘤活性,与deruxtecan为基础的ADC相比,其表现出更强的旁观者杀伤效应,这要归功于与DXd相比,exatecan的被动细胞渗透性改善。使用这种药物-连接子的策略已开发了两种ADC药物:PRO1184和PRO1160 , DAR均为8。
最近的体内研究也表明,exatecan不需要氟环功能来发挥其抗肿瘤活性,从而扩大了分子的官能化以产生可连接的衍生物。使用这种策略开发的最有前途的ADC(mAbE21a,derivative 11, DAR7.5)在EGFR+模型中显示出优异的抗肿瘤活性,在0.25 mg/kg时即可完全缓解。
一种新的特有exatecan衍生物AZ’0132最近被披露,目前已作为ADC 药物AZD8205的有效载荷进行I/II期临床研究(NCT05123482),靶标是B7-H4(图3)。
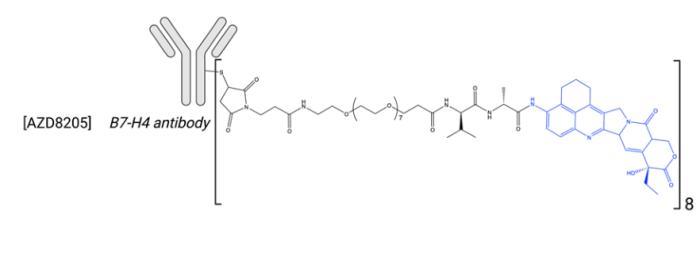
图3. AZD8205结构示意图
Irinotecan
Irinotecan已被FDA批准用于治疗各种实体肿瘤,如胃肠道恶性肿瘤、胶质母细胞瘤和宫颈癌,是拓扑异构酶1抑制剂SN-38的前体药物。SN-38不溶于水,会导致严重的毒性,包括强烈的骨髓抑制和重度腹泻。因此,开发Irinotecan是为了提高生物利用度并获得可接受的治疗指数。
IMMU-132(Trodelvy®)是一种TROP2抗体与基于SN-38的药物连接子相偶联的ADC药物。该ADC已于2020年被FDA批准用于治疗三阴性转移性乳腺癌和转移性尿路上皮癌,目前正在进行治疗HR+/HER2-、前列腺癌和子宫内膜癌的临床试验(NCT03725761和NCT04251416)。使用这种基于SN-38的连接子也已经开发了其他ADC药物,包括分别以CEACAM5和HLA-DR为靶点的IMMU-130和IMMU-140(图4)。
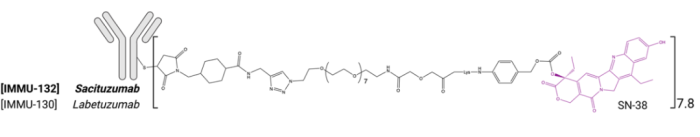
图4. 基于SN-38的代表ADC药物结构示意图
最近,A7R-SN-38 ADC已被开发用于治疗自身免疫性疾病,以避免类固醇耐药性。
Belotecan 衍生物
另一种拓扑异构酶1抑制剂KL610023是FDA批准的Belotecan分子的衍生物,正在作为ADC有效载荷进行研究。基于这种拓扑异构酶I抑制剂开发了抗TROP2 ADC药物SKB-264,DAR为7.4(图5)。目前正处于I/II期临床试验(NCT04152499)中,用于治疗各种实体肿瘤患者。
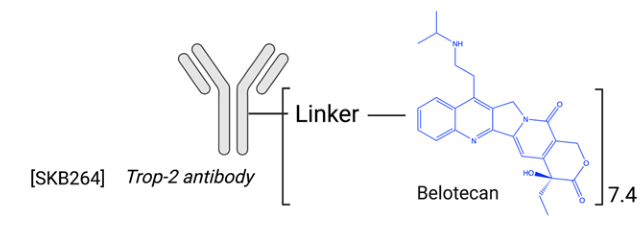
图5.SKB264结构示意图
其他拓扑异构酶1抑制剂
以喜树碱为基础的衍生物作为ADC有效载荷的限制之一是分子中缺乏可连接的化学胺基团。已经合成了其他CPT衍生物,以在不改变其抗肿瘤特性的情况下在有效载荷内插入可连接的功能团。在这些衍生物中,cAC10是一种CD30抗体与8个AMDCPT分子连接的ADC药物,临床前研究显示出优异的效果有希望的结果。
最近已经开发了几个非CPT衍生物,包括吲哚异喹啉,二苯并萘酮和氟吲哚异喹啉(图6)。与CPT衍生物相比,这些分子表现出几个优点,包括更高的细胞毒性、更好的稳定性或更长的活性,目前正处于早期临床试验阶段。
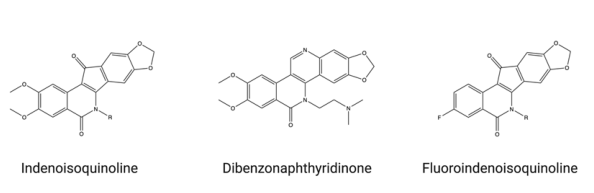
图6. 下一代拓扑异构酶I抑制剂作为ADC的潜在有效载荷
已进入临床试验的其他有效载荷:希望与挑战并存
虽然拓扑异构酶1抑制剂已经深刻地改变了ADC的有效载荷格局,但其他一些有效载荷已经在临床试验中进行了评估,主要包括拓扑异构酶2抑制剂、RNA聚合酶抑制剂、Bcl-xL抑制剂和免疫刺激剂。
拓扑异构酶2抑制剂
拓扑异构酶2抑制剂广泛应用于血液系统恶性肿瘤和实体瘤的抗癌治疗。它们的作用机制复杂,不仅可能直接抑制拓扑异构酶2的活性,还可能涉及DNA嵌入、ROS诱导和线粒体破坏。它们的毒性包括骨髓抑制、胃肠道毒性,在某些情况下还会出现严重的心脏毒性。
目前已有多种拓扑异构酶2抑制剂ADC药物进入临床阶段(图7)。第一个含有拓扑异构酶2抑制剂的ADC 药物SGN-15(BMS-182248)是由阿霉素偶联到小鼠BR-96抗体上,靶向Le-Y抗原。其I期临床试验显示出可接受的耐受性,但II期由于连接子的不稳定和Le-Y靶标在正常组织中的表达而导致靶外毒性而终止。阿霉素后来被连接到CD74靶向抗体Milatuzumab(IMMU-110)上,用于治疗多发性骨髓瘤。但疗临床效令人失望,于2013年停止(NCT01101594)。
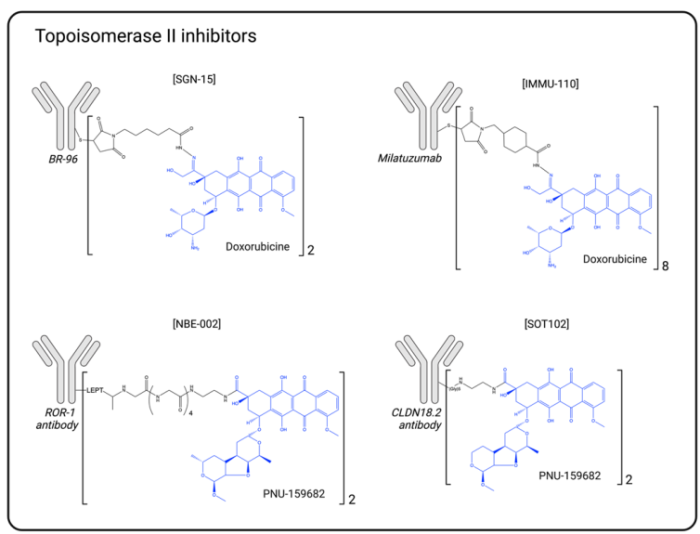
图7. 拓扑异构酶2抑制剂示意图
考虑到阿霉素作为ADC有效载荷的局限性,后来又开发了另一种细胞毒性比阿霉素高100倍的蒽环类药物PNU-159682。除了比其他拓扑异构酶2抑制剂更有效之外,PNU-159682也不是外排底物。2020年,一种基于PNU-159682的新型ADC 药物NBE-002,靶向ROR1,进入了I/II期临床试验(NCT04441099)。另外,NBE-002还诱导了长期的免疫保护,这使得它可以成功地与免疫检查点抑制剂组合治疗。
SOT102是另一款基于PNU-159682的ADC药物,靶标是CLDN18.2。SOT102在低表达肿瘤中显示出很大的治疗窗口,并已于2022年4月进入I期临床试验。
转录抑制物
转录在细胞发育、活性和增殖中起着重要作用,因此可以构成ADC有效载荷的创新和原始靶点。转录由直接与DNA结合的RNA聚合酶II(RNApolII)调控,涉及与RNApolII形成复合体启动转录(如TFIIH)的转录因子和调节染色质结构和可及性的辅助调节因子(如组蛋白脱乙酰酶,HDAC)。虽然一些HDAC抑制剂已获得批准,但由于RNApolII抑制剂耐受性较差,目前还没有批准。
Amanitin是一种天然的、高效的RNApolII抑制剂,来源于鹅膏菌。α-Amanitin和β-Amanitin与其他七个大环衍生物一起构成鹅膏毒素家族。尽管它们被广泛用作实验室试剂来探索转录机制,但事实证明,α-Amanitin毒性太大,特别是对肝脏,不能作为抗癌剂进一步开发。然而,这种分子作为一种潜在的ADC有效载荷呈现出许多优点,包括细胞内靶点、良好的物理化学性质(包括亲水性)、它对外排泵的不敏感性,以及它在静止的癌细胞中产生细胞毒性的能力。β-Amanitin于1973年首次与白蛋白偶联,显示出对巨噬细胞的选择性杀伤。该衍生物后来与抗MUC1和抗PSMA抗体偶联,并在临床前模型中显示出强烈的选择性细胞毒性。
2021年5月,首个Amanitin抗体偶联物(ATAC®) HDP-101进入早期临床试验(图8)。HDP-101是一种针对BCMA的ADC,目前正在多发性骨髓瘤和浆细胞疾病患者中进行评估(NCT04879043)。ATAC最近被发现可以是一种免疫激活药物,它们被发现可以诱导免疫原性细胞死亡(ICD),并与ICI表现出协同作用,这为临床环境中联合应用的可能性开辟了新的视野。许多针对其他靶点(EpCAM、HER2、PSMA、CD19)的α-Amanitin ADC在体外和体内也都已显示出强大的抗肿瘤活性。
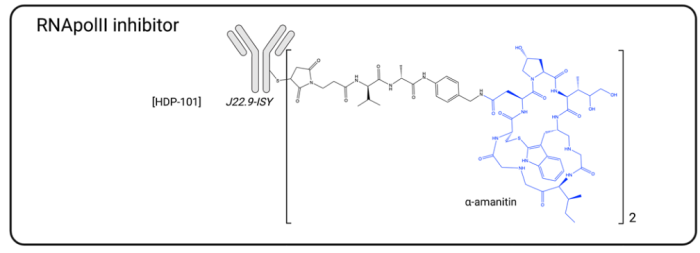
图8. HDP-101结构示意图
其他高效的RNApolII抑制剂在20世纪90年代已被偶联应用,如鬼臼毒素,霉菌毒素毛霉烯、疣草素A和大蒜素A。这些化合物在各种细胞系中具有纳摩尔细胞毒性,有望是未来几年进一步研究的对象。
另一种阻止DNA转录的策略是抑制转录因子(TFs)。TF对于RNApolII在起始步骤中与DNA的连接至关重要。TF抑制剂(TFi)已经在水溶性前药minnelide的临床试验中证明了其抗肿瘤活性,目前处于II期评估(NCT04896073)。雷公藤甲素是一种来源于中草药“雷神藤”的天然化合物,具有很高的细胞毒性,但也是疏水性的,生物利用度低,毒性高。因此,正在努力开发具有更好药物化学性质的类似物。另一种策略是将这种分子与靶向实体偶联,从而绕过这些问题。雷公藤甲素最近首次与CD26抗体偶联靶向间皮瘤和淋巴瘤,这种不可切割的ADC有效地阻止了靶细胞中mRNA的合成,并表现出优异的体外和体内抗肿瘤活性。
HDACs(组蛋白脱乙酰酶)影响转录因子,因此参与包括转录在内的各种细胞过程。已发现它们在癌细胞中过度表达或过度活化,并被认为与增殖、迁移和侵袭的增加有关。Vorinostat和dacinostat是FDA批准的HDAC抑制剂(HDACi)的两个例子。然而,这些分子存在很大的全身副作用风险,如血小板减少症和胃肠道毒性,以及较差的PK特性。自2018年以来,它们一直在ADC设计中进行研究:ST74612AA1是第一个生物偶联HDAC抑制剂。这种相对无毒的分子是第二代pan-HDACi。这种分子已与西妥昔单抗和曲妥珠单抗成功偶联,产生的两种ADC都比未偶联的HDACi更安全,同时在CDX和PDX模型中均具有活性。2020年,vorinostat和dacinostat也与西妥昔单抗和曲妥珠单抗偶联,并在体外显示抗肿瘤活性。
Bcl-xL抑制剂
Bcl-2家族成员可以是pro (Bad,Bim,PUMA,Bik,Bak,Bax)或抗凋亡蛋白(Bcl-2、Bcl-xL、Bcl-w、Mcl-1)。在癌细胞中,这些蛋白质之间的平衡更通常倾向于存活,使得抗凋亡蛋白质成为创新ADC有效载荷的靶标。
Bcl-xL和Bcl-2抑制剂根据其化学功能分为4大类:N-酰基磺酰胺类(navitoclax,venetoclax),吲哚类(obatoclax),醋酸棉酚(AT-101,sabutoclax)和苯并噻唑腙类(如WEHI-539)。Bcl-xL的抑制与严重的血小板减少症有关,证明了寻找高度特异性的Bcl-2抑制剂如venetoclax的必要性。目前,venetoclax被批准用于慢性淋巴细胞性白血病和急性髓细胞性白血病患者。
ABBV-155 (mirzotamab clezutoclax)是B7-H3抗体与Bcl-xL抑制剂clezutoclax偶联的创新ADC药物,其在2018年进入了I/II期临床试验,用于治疗晚期非小细胞肺癌和乳腺癌患者的晚期实体肿瘤(NCT03595059)。单药I期队列中的前31名患者中未报告剂量限制性毒性,严重副作用包括贫血、淋巴细胞数减少、疲劳和腹泻。21%的患者在紫杉醇联合治疗组中观察到部分反应。
激酶抑制剂
人类基因组包含超过500种激酶,其中超过150种与包括癌症在内的各种疾病相关。蛋白激酶是催化磷酸化的酶,分为3类:丝氨酸、苏氨酸或酪氨酸激酶。在癌症中,多种激酶家族参与细胞周期进程、细胞增殖、运动和血管生成。
虽然蛋白激酶抑制剂在癌症治疗方面得到了广泛研究,但作为ADC的有效载荷尚未得到广泛研究,可能是因为它们的效力较低。抗CD19抗体B43已经与木黄酮偶联,发现木黄酮可通过抑制酪氨酸激酶受体表皮生长因子受体(EGFR)从而诱导细胞凋亡和细胞增殖抑制。体外和体内临床前研究表明,累积剂量为100 mg/kg时无毒性,且在小鼠模型中比标准化疗具有更强的抗肿瘤效果。这些有优异的结果使得其在1999年对ALL和NHL患者进行了临床研究。其在人体内呈现良好的药代动力学特征外,没有毒性,且有优异的抗肿瘤活性。然而,这种ADC药物的进展还没有进一步的报告(NCT00004858)。
最近,其他激酶抑制剂作为ADC有效载荷也进行了评估。包括neolymphostin (一种PIKK抑制剂),以及dasatinib 和staurosporine(多激酶抑制剂)。但总体而言,ADC形式的酪氨酸激酶抑制剂的疗效目前被发现是有限的,该家族很难在晚期患者中获得成功。
免疫刺激抗体偶联物
针对适应性免疫系统的免疫检查点抑制剂的成功极大地刺激了研究人员利用先天免疫系统刺激的抗肿瘤研究。然而,最有效的药物如STING和TLR激动剂的全身给药与严重的全身毒性有关,这是由细胞因子释放综合征引起的,从而将当前的研究限制在肿瘤内注射。因此,它们与抗体的结合似乎是一种在改善耐受性的同时开发其强大的抗肿瘤潜力的有希望的手段。
免疫刺激抗体偶联物代表了一种新的抗体-药物偶联物,目前有两个ADC已进行临床试验 (NJH395,BDC1001),另外一个SBT6050其临床评估因战略决定而终止 (图9)。
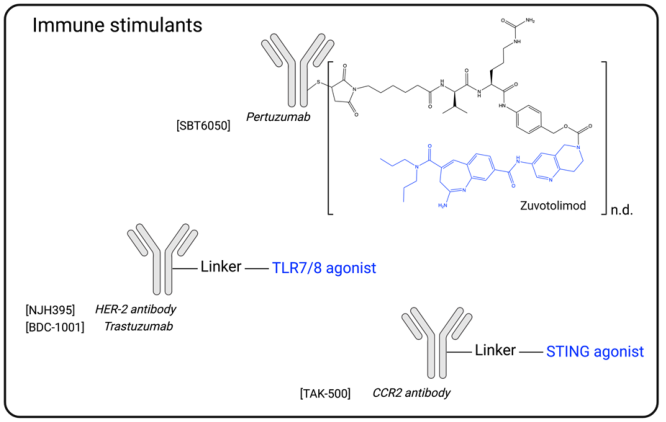
图9. 免疫刺激抗体偶联物结构示意图
小编小结
有效载荷是ADC的重要组成部分,在ADC领域的未来发展中,有效载荷多样化将发挥关键作用,其有望将ADC的治疗武器库开放给其他尚未受益于靶向治疗的癌症。
开发这些新型有效载荷的一个关键问题是减轻它们的副作用。目前批准的ADC已表明,它们具有预期的(骨髓抑制、神经毒性)或意想不到的(如眼或肺)毒性。因此,获得令人满意的治疗指数将是创新ADC有效载荷未来发展的关键。
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多