ADC的优化之路:道阻且长,行则将至
一、靶点
目前在ADC中能利用的靶抗原或受体范围很广。大多数靶点是能通过受体介导的内吞作用(RME)内化的分子,但也有少数靶点存在于细胞表面或肿瘤血管系统中。对于这些内化靶点,设计ADC的最佳条件是让它们在肿瘤细胞上丰富、均匀地表达,在健康的组织和器官中不表达或低表达。然后在ADC结合后迅速内化,并传递到溶酶体。
表1 临床ADC总结
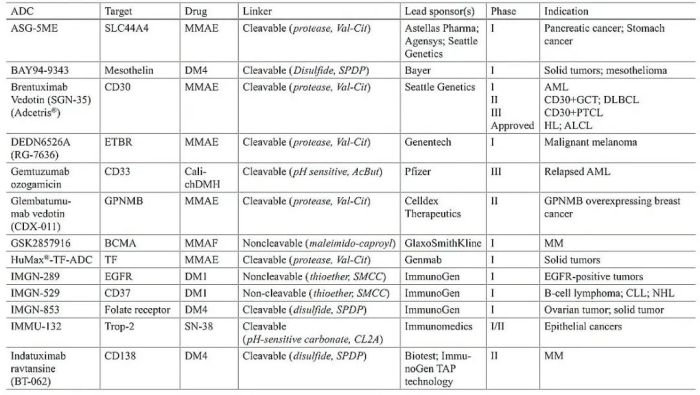
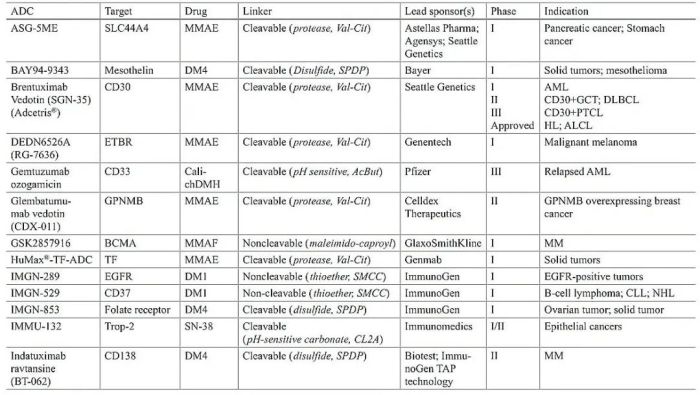
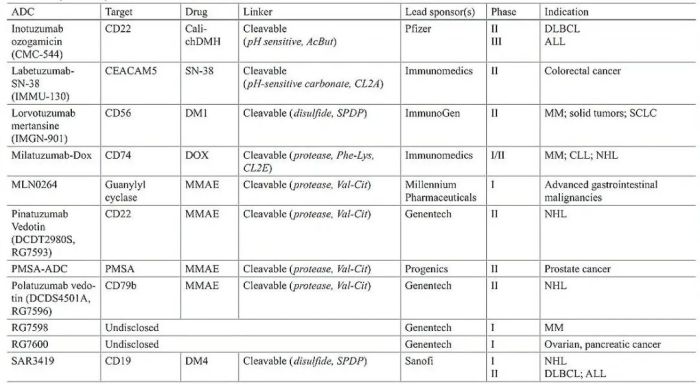
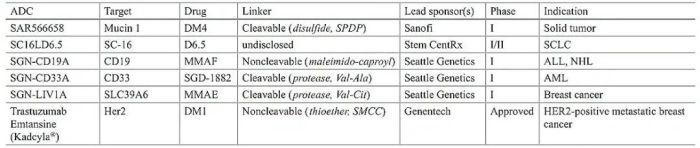
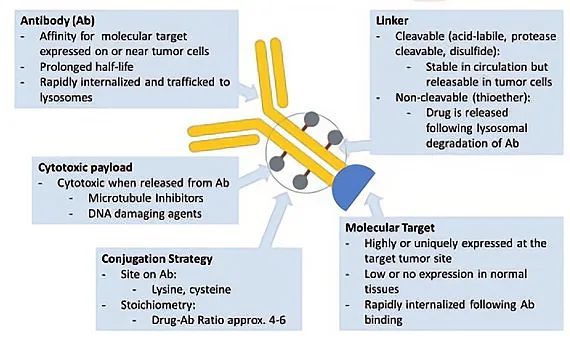
图1 ADC结构。设计ADC的主要考虑因素包括分子靶点、抗体、化学结合、细胞毒性有效载荷以及抗体和细胞毒性有效载荷之间的连接物。
CD30完美符合这一标准。CD30在肿瘤细胞中丰富均匀的表达,在正常组织表达量较低。和抗体结合后迅速内化。FDA批准的Adcetris®就是靶向CD33的ADC。但不是所有ADC都能完美符合这一条件。
如Kadcyla®的靶点HER2虽然能在乳腺癌组织中高度表达,并在抗体结合后迅速内化。但它也会在皮肤、心脏、胃肠道(GI)等其他组织中表达,但HER2在器官中表达还是低于靶肿瘤细胞,因此Kadcyla®仍然具有良好的耐受性。
在临床开发中比较ADC时,大部分差异性是由靶点带来的,因靶点不同,抗体设计就不会不一样。靶点的范围很广,如:
①肿瘤细胞表面抗原(例如,Kadcyla®中的HER2)
②表面受体,如上皮生长因子受体(EGFR;IMGN-289)和叶酸受体(IMGN-853),
③细胞黏附分子,如Nectin-4(ASG-22ME)
④糖蛋白,如粘蛋白1(SAR566658)和GPNMB(CDX-011)。
阻碍靶点开发的关键问题在于肿瘤抗原密度和异质性,以及正常组织和器官中可接受的表达水平。研究表明,ADC治疗效果与抗原密度相关;但最近研究表明,表达水平可能不是ADC疗效的决定因素,对抗CD79b ADC(DCDS4501a)的研究表明,在CD79b表达水平高于最低阈值的细胞系中,表达水平与体外活性存在非线性关系。因此,可能存在一个最小阈值,不同靶点的阈值可能不同。
对于健康组织的脱靶效应,需要考虑该靶点是否能在重要器官或再生组织中表达,以及ADC的可及性。
例如在骨髓中表达,就意味着毒性可控。因为在大多数情况下B细胞和髓系细胞都可以再生。
PMSA也是一个典型例子,虽然它在癌变和健康的前列腺组织中都有表达。但由于大多数前列腺癌患者会选择切除前列腺,ADC的毒性载荷对前列腺影响并不会特别严重。
PMSA在肾脏和小肠的顶膜上也有表达。但事实上,被限制在基底外侧的ADC很难接触到细胞顶膜,因此毒副作用带来的影响也不会特别大。
由此可得,深入了解分子靶点的表达、特异性和疗效、副作用之间的关系将有助于ADC的未来发展。
二、抗体设计
抗体成分是ADC靶向效率和药代动力学的重要决定因素。ADC抗体的大小、转运机制、抗体效应功能以及和Fc受体结合的能力都是ADC设计的考虑因素。ADC通常具有与亲本抗体类似的低清除度和长半衰期。但由于药物-抗体比率(DAR)和药物切割的额外代谢途径,ADC的半衰期和清除率可能与亲本抗体成分略有不同。
抗体设计的考虑因素之一是它的亚型(IgG1、IgG2、IgG3和IgG4),由于Fc区域的差异,各亚型效应也可能不同。在抗体依赖的细胞介导的细胞毒性(ADCC)和补体依赖的细胞毒性(CDC)中,IgG1和IgG3亚型通常更活跃,而IgG4往往缺乏效应功能。虽然参与这些次级免疫功能有利于抗肿瘤活性,但可能会影响肿瘤定位。
IgG1是ADC中最常见的亚型,用于ADCC效应较强的Kadcyla®和Adcetris®中。使用IgG4亚型的Mylotarg®没有ADCC或CDC效应。
使用较小抗体片段,是未来对ADC抗体成分的改进的一种思路。如体积较小的纳米抗体,具有更高的肿瘤渗透率。第二种改善抗体功能的方法,是能修饰特定位点的工程策略。
三、靶点
因为连接位点和DAR都会影响PK/PD、结合亲和力和聚集状态,它们在ADC开发中至关重要。临床上大多数ADC利用赖氨酸的ε氨基或半胱氨酸的巯基。在赖氨酸参与修饰时,药物分子分布在抗体中的许多赖氨酸残基上,导致DAR范围不同。
半胱氨酸残基修饰与抗体铰链区二硫键的减少有关,并会出现偶数的DAR。Kadcyla®(每个抗体3.5个DM1分子)和Adcetris®(每个抗体~4个MMAE分子)分别通过赖氨酸修饰和半胱氨酸偶联生成。
虽然与抗体结合的药物分子结合,意味着越多的有效载荷,但它也会导致终产物聚集度更高,pk和结合亲和力都低于亲本抗体。在药物偶联的情况下,ADC的半衰期往往比未修饰的抗体短。因此,为了保持较长的半衰期和较高的抗体结合效率,平均DAR通常保持在3-4左右。
当前化学偶联策略还有一个主要缺点是会在不同位置产生具有多个DAR的不均匀终产物。计算可得,通过修饰赖氨酸残基可以将药物分子分布到大约40个不同的位置,从而产生>106个不同的ADC种类,通过半胱氨酸残基的结合可以减少异质性,但仍然会产生超过100种ADC。
为了克服这一问题,几家不同的公司正尝试对特定位点进行修饰来产生均质ADC,主要技术涉及使用工程半胱氨酸残基、加入非天然氨基酸、以及酶偶联,如下所述。
01·
工程半胱氨酸残基
位点特异性修饰的策略之一是通过单点突变将半胱氨酸残基引入抗体序列。这项由基因技术公司开发的技术,称为ThioMAb。首先利用噬菌体展示技术来选择合适的连接位点,再通过单点突变将半胱氨酸残基(通常n=2)引入抗体氨基酸序列。最后通过马来酰亚胺连接子连接到工程半胱氨酸上。
02·
加入非天然氨基酸
Sutro Biophma和Ambryx公司,正在使用基于非天然氨基酸的方法来定点修饰ADC。在这种方法中,天然氨基酸中不存在可以结合或使用生物正交化学方法进行特异性修饰的独特官能团,这些技术克服了半胱氨酸修饰的马来酰亚胺和二硫键交换化学的局限性,不进一步酶处理步骤。Ambryx引入了一个对乙酰苯丙氨酸基团(PAcPhe),该基团含有一个酮基,可以选择性地偶联到烷氧基胺类药物上,形成稳定的肟键连接子。Sutro Biophma技术引入了一个p-叠氮甲基-l-苯丙氨酸(PAMF)基团,该基团可以通过菌种促进的叠氮-炔环加成(SPAAC)点击化学与药物分子反应。
四、偶联
Innate Pharma正在利用酶促翻译后修饰来实现精确的位点特异性结合。这项技术需要从抗体中去除N连接的多糖,或者使用通过定点突变产生突变的糖基化变体。随后,使用细菌转谷氨酰胺酶(BTGase)将缺乏cadaverine的药物底物连接到抗体上,进行位点特异性连接。还有许多其他特定部位的技术也在逐渐开发,比如与天然糖链残基的结合(SynAffix的GlycoConnectTM)和Medtope Biosciences的“Medtope-Enabling”技术等。
五、细胞毒性有效载荷
ADC中使用的有效载荷主要有两类,一是破坏微管组装并在有丝分裂中发挥重要作用的抗分裂药物。以及与DNA小沟结合导致DNA双链断裂的药物 (图2)。
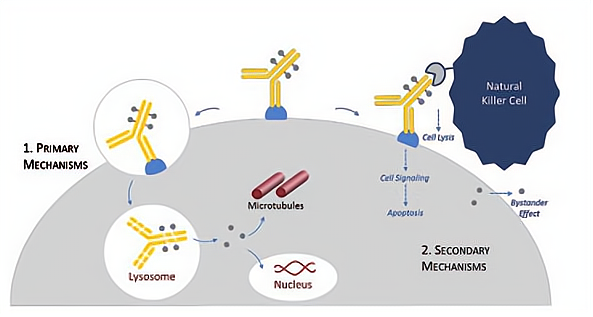
图2 ADC内化及其毒性机制的研究进展。在与细胞表面受体结合后,ADC被受体介导的内吞作用内化。ADC被受体介导的内吞作用内化。该细胞毒性药物通过连接物的裂解或抗体的降解而释放,然后发挥其细胞毒性。
抗有丝分裂药物MMAE和DM1/DM4是最常用的药物(表1,图3),而且由于它们优先破坏高增殖细胞和增加恶性细胞对有丝分裂的敏感性,还具有额外的特异性。与阿霉素和紫杉醇等传统化疗药物相比,这两类ADC药物效力都要高得多(100-1000倍)。
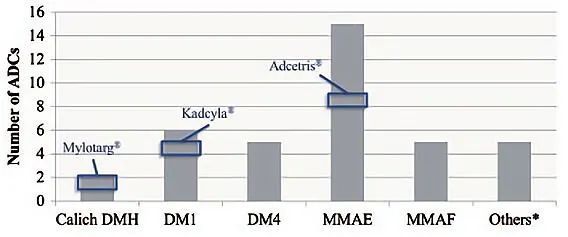
图3 ADC临床用药情况比较。
虽然ADC主要靶向肿瘤部位的,但据统计,只有1-2%的给药剂量会聚集在靶点。因此,提高药效应是ADC开发的重点。
尽管ADC的抗原/抗体和连接子有很大的变异性,但使用的药物类型有限。开发适合ADC的新药是一项艰巨的任务。首先,药物需要有很高的细胞毒性。其次,药物靶点应在细胞内。同时它的尺寸应该很小,以减少免疫原性的风险和水的溶解性问题;它必须在循环中稳定;它应该有一个与抗体药物连接的连接子。因此,在识别适用于ADC的药物上仍有很大的开发空间。
结 语
经过几十年的试验和研究,ADC已成为了最新的癌症治疗药物类别。随着两种已批准的药物在临床上表现良好,以及30多种处于不同临床研究阶段的药物,ADC药物发现和开发的前景非常光明。有效载荷、靶向抗体和连接子技术的优化有利于产生具有更高的药物输送效率、更少不良反应特征,和减少耐药性和癌症转移可能的下一代ADC。
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多