人工智能中的药物设计:应用与趋势

图1. 全球明确使用人工智能的医药公司中的十大药企
人工智能(AI),特别是其中的深度学习,为创新药物的设计提供了机会。近几年出现了各种机器学习方法,其中一些被认为是特定领域的人工智能实例,并且许多人工智能已成功地用于药物设计。
本文先概述了4个有代表性的人工智能在药物设计中的应用,包括三种药物设计方法以及药物构效关系解释,接着讨论了人工智能中的药物设计趋势,希望可以为各位读者提供药物设计潜在未来方向的参考。
1
针对靶标蛋白的新药设计
近年来,基于深度学习的方法成为新药设计的一种很有前途的工具。这些方法大多是基于配体的,其中初始的靶标特定的配体数据集,对于设计具有优化性质的有效分子是必要的。尽管已经有人尝试开发另一种方法来设计靶标特定的配体数据集,但在设计针对新靶标蛋白的分子时,此类数据集的可用性仍然是一个挑战。
Sowmya, Navneet[4]等人提出了针对靶标蛋白活性部位的深度学习方法。首先,使用图形注意力模型来学习实验上已知的形成蛋白质−配体复合体的蛋白质活性部位,了解其氨基酸的结构和特征。接下来,将学习到的活性中心特征与预先训练的生成模型一起用于有条件地生成新分子。然后在强化学习框架中使用生物活性预测模型,来优化条件生成模型(如图2所示)。
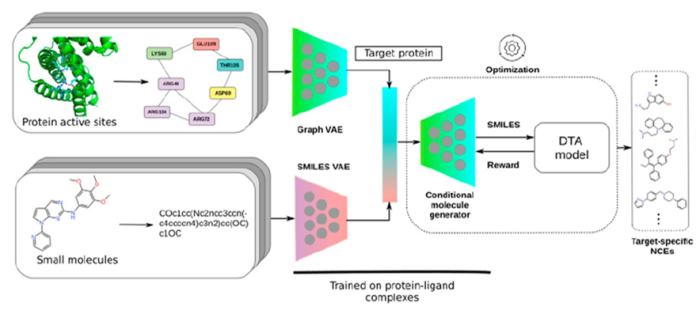
图2. 基于蛋白质结构的药物设计流程,来源:参考文献4
最后,用两种蛋白质Janus kinase2(JAK2)和多巴胺受体D2(DRD2) 验证了方法,在这两种蛋白质中,产生了与已知抑制剂类似的分子。图形注意力模型可以识别可能的关键活性部位残基,从而影响条件分子生成器设计具有与已知抑制剂相似的药理特征的新分子。
2
基于配对多目标优化的新药设计
多目标优化解决了药物的自动化从头设计问题,这是人类创造过程的结果。Alberga, Gambacorta[5]等人设计了一种新的配对多目标方法,该方法在基于递归神经网络的自适应生成算法中实现,用于自动从头设计新药物分子,其总体特征通过在相关物理化学性质 (MW、logP、HBA、HBD) 和偏向特定生物靶标的约束之间找到最佳权衡来优化。
他们进行了针对SARS-CoV-2主要蛋白酶、乙酰胆碱酯酶、神经氨酸苷酶的药物分子的从头设计。采用几种质量指标(包括有效性、唯一性、内部多样性、过滤值、综合可达性)来评价药物的相似性、化学可行性、多样性和有效性。最后进行了分子对接,以进一步评估新生成的抑制剂对相应所需生物靶标的有效性情况(如图3所示)。
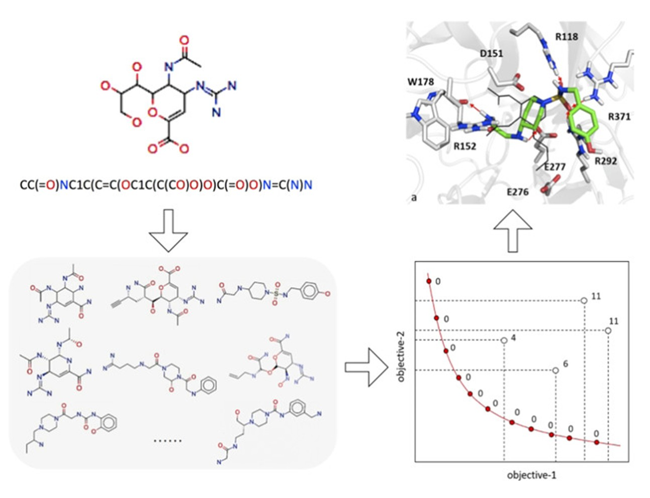
图3. 多目标优化方法流程图,来源:参考文献5
结果表明,多目标优化能够捕捉连接化学和生物方面的潜在联系,从而为可定制的设计策略提供易于使用的选择,这对线索生成和线索优化都特别有效。
3
在确定了一个靶点后,寻找苗头化合物是基于结构的药物设计的重要第一步,常用的方法有虚拟筛选、组合化学、高通量筛选、DNA编码化合物库筛选等。Minsup, Kichul[6]等人设计了一种靶向特定的药物设计方法,该方法利用了基于深度学习算法和水药效团的seq2seq模型。
首先通过深度学习算法,设计出一系列满足类药五原则等诸多性质的苗头化合物,接着使用分子对接和药效团模型,评估这些苗头化合物的有效性,其中的药效团模型用的是水药效团模型,这个模型通过水分子的分子动力学模拟来构建药效团特征,其中药物靶点的水药效团特征如图4所示。
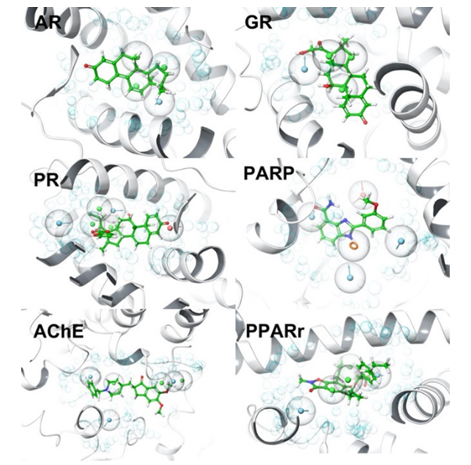
该方法可以自主产生一系列靶向有利的化合物,从大规模的化合物库中筛选出匹配药效团特征的分子,以这些分子作为输入,训练分子生成模型。最后用药效团筛选生成的分子,得到一批候选化合物。水药效团模型用于在大型化合物数据库中,筛选对给定目标有利的化合物,而seq2seq化合物生成器用于训练筛选出的化合物,并基于训练模型生成全新的化合物。
4
解释药物中的构效关系
DNN指的是由神经网络组成的、复杂的非线性统计模型[10],该网络具有多个用于预测的隐含层(如图5所示)。更快的计算机硬件、改进的算法,以避免神经网络过度匹配,再加上许多计算机平台上软件解决方案的可用性以及改进的训练算法,推动了 DNN在人工智能领域的成功应用,例如计算机视觉、语言处理以及药物设计。
在计算机领域,基于深度神经网络 (DNN) 的模型在预测新分子的活性和性质方面很有前途。不幸的是,它们固有的黑箱特征阻碍了我们对分子结构作用的理解。然而,这些信息对于研究构效关系(SAR) 以指导进一步优化至关重要。
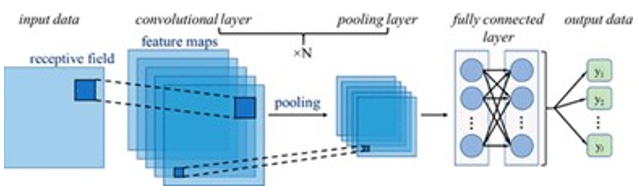
图5. 卷积神经网络 (CNN) 示意图,来源:参考文献10
为了解决这一解释差异,“可解释人工智能”(XAI) 方法最近变得流行起来。Tobias, Hans[7]等人将多种XAI方法应用于SAR和X射线晶体结构的先导优化数据集的项目(如图 6 所示),并进行了比较,结果说明将DNN模型与一些强大的解释方法相结合,可以得到易于理解的、全面的解释。
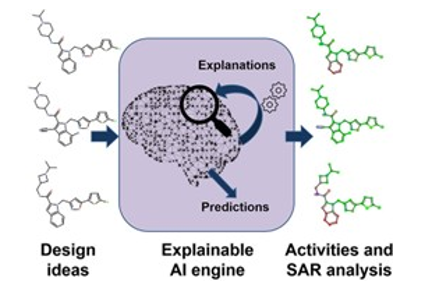
图6. 解释构效关系的方法流程,来源:参考文献7
这些解释可以使用热图以一种全面的方式收集和呈现,以清楚地突出分子的哪些部分被认为对解释亲和力或任何其他目标性质有利或不利,这将激发计算化学家的创造力,以设计出具有前瞻性思维的药物。
5
未来改进趋势
方向合理的药物设计方法正在不断改进,这些方法正在接近更广泛的药物靶点,未来还可以预计会有各种各样的额外改进:
1. 改进的计算机硬件允许使用更严格的方法应用于大分子系统,如果在未来看到量子力学对接研究的出现也并不令人惊讶,此外还会促进混合方法的发展[2](遗传神经网络、近邻遗传算法等)和药物设计集成工具(设计与评估一体化)的开发。
2. 基于人工智能的蛋白质靶点结构、位点解析将会更加深入地渗透进药物设计中,即蛋白质结构建模的进展,促进了基于结构的药物设计在未结晶的药物靶点上的广泛使用,它们是化学设计的补充,并且在选择用于风险评估的实验系统时很重要。例如:AO底物类药物的设计[3](AO是醛氧化酶,结构作用如图7所示,包括催化氮杂杂环和醛的氧化、酰胺水解和多种还原),AO底物在上市药物中很少见,许多候选药物由于药代动力学活力差、种间差异和不良反应而失败。
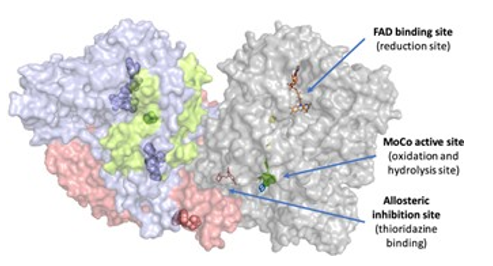
图7. 醛氧化酶的结构及其结合位点,来源:参考文献3
由于大多数问题源于复杂且知之甚少的AO生物学结构,因此有效的解决方案是停止或减少AO代谢:早期考虑AO介导的代谢(例如,在苗头化合物和先导化合物优化中),但不是主动避免AO结构,其中许多是有价值的和常见的构建块(例如,各种氮杂杂环和酰胺),及时合理地应用药物设计来控制AO介导的代谢(例如,停止、减少或用于前药设计);
3. 与人工智能相结合的新实验方法将药物设计引向新的方向,有许多类似的例子:如果没有固相合成方法,组合化学[11](实例如图 8 所示)和高通量筛选将不会像今天这样有用;在催化剂设计等领域的改进,在催化剂设计等领域的改进,使我们可以快速访问更多的化学结构,使生物活性测定可以利用更广泛的生物靶标。
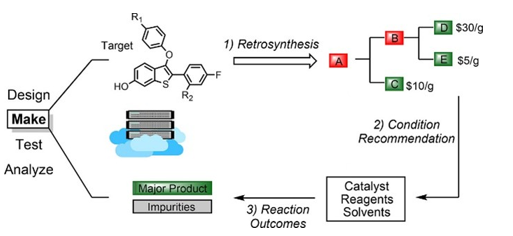
图8. 组合化学实例,来源:参考文献11
例如,结合人工智能和统计学方法对CADD的药物评估算法本身进行优化[8],定量方法仅对结构相似分子的抑制剂之间的相对结合亲和力提供准确预测,而定性方法为结构更多样化的一组化合物的相对结合亲和力提供定性趋势。
理想情况下,结合这两个特征将大大提高药物评估算法在药物设计中的效用(如图9所示)。即结合人工智能和统计学方法计算出包含上述许多特性的回归方程,然后将方程嵌入CADD的药物评估算法,将极大有利于药物设计。
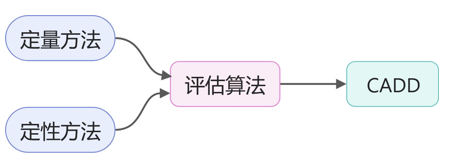
图9. 定性、定量方法对CADD的影响
本文来源于药渡
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多