自微乳化在多肽口服中的应用——是希望耀光还是无味冷汤
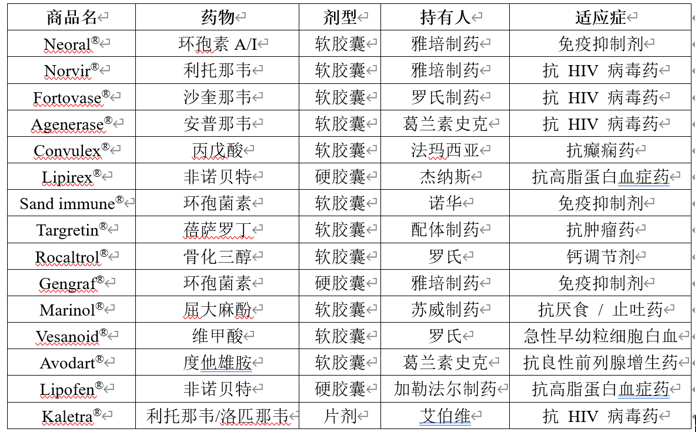
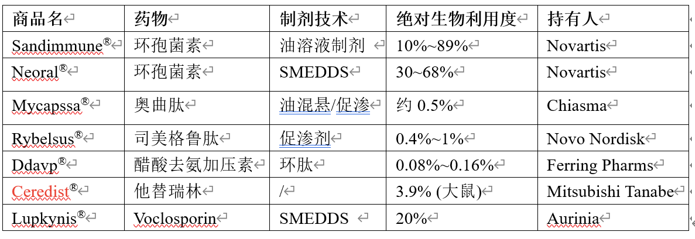


参考文献:
1、Pehlivanov I. Self-emulsifying drug delivery systems as an approach to improve therapeutic effectiveness of orally administrated drugs. J of IMAB 2019; 25 (2):2575-82.
2、Jaiswal P, Aggarwal G. bioavailability enhancement of poorly soluble drugs by SMEDDS: a review, Journal of drug delivery and therapeutics, 2013; 3(1):98-109.
3、Harvey JH, McFadden M, Andrews WG, Byrne PJ, Ahlgren JD, Wolley PV. Phase I study of echinomycin administered on an intermittent bolus schedule. Cancer Treat Rep 1985, 69, 1365-1368.
4、Heejun Park, Eun-Sol Ha, Min-Soo Kim. Current Status of Supersaturable Self-Emulsifying Drug Delivery Systems. Pharmaceutics. 2020, Vol.12, No.4, p.365.
5、I D van Mourik , M Thomson, D A Kelly. Comparison of pharmacokinetics of Neoral and Sandimmune in stable pediatric liver transplant recipients. Liver Transpl Surg.1999 Mar;5(2):107-11.
6、Khoo SM, Porter CJ, Edwards GA, Charman WN.Intestinal lymphatic transport is the primary route for halofantrine after oral postprandial administration. Pharm Sci ,1999; 1: 624
7、Subramanian D.A., Langer R., Traverso G. Mucus interaction to improve gastrointestinal retention and pharmacokinetics of orally administered nano-drug delivery systems. J. Nanobiotechnol. 2022;20:362.
8、DS Nielsen, NE Shepherd, W Xu, AJ Lucke, MJ Stoermer, DP Fairlie. Orally Absorbed Cyclic Peptides.Chemical reviews 117 (12), 8094-8128.
9、A. Mllertz, A.Ogbonna,S. Ren,T. RadesNew perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs.J. Pharm. Pharmacol.62 (2010), 1622-1636.
药事纵横
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多