一文带你了解外用半固体制剂IVRT
要想在研发过程中始终保证药品质量,研发人员必须关注产品的关键质量属性(CQAs)。针对半固体制剂而言,其关键质量属性包括外观、混悬药物的晶型、粒度分布、液滴粒径、流变特性、 pH值、黏度、含量均匀度、微生物限度、有关物质、抑菌剂含量及抗氧剂含量、无菌(用于烧伤(除轻度 I°或 II°外)或严重创伤的无菌制剂)以及体外释放试验( IVRT)和体外透皮试验( IVPT) 等。
前言
要想在研发过程中始终保证药品质量,研发人员必须关注产品的关键质量属性(CQAs)。针对半固体制剂而言,其关键质量属性包括外观、混悬药物的晶型、粒度分布、液滴粒径、流变特性、 pH值、黏度、含量均匀度、微生物限度、有关物质、抑菌剂含量及抗氧剂含量、无菌(用于烧伤(除轻度 I°或 II°外)或严重创伤的无菌制剂)以及体外释放试验( IVRT)和体外透皮试验( IVPT) 等。其中流变特性、体外释放试验( IVRT)以及体外透皮试验( IVPT)属于半固体制剂药学研究过程中具有代表性的重要技术方法。本文主要介绍IVRT在半固体制剂研究中的地位及其方法开发过程中的注意事项。
IVRT的地位
在药品研发过程中,临床试验是证明药品的有效性的有力证据。但是,临床试验高昂的时间和资金成本使其不适合作为药品质控的常规方法。因此,必须寻找体外测试方法代替临床试验来保证研发过程中药品质量的一致性。通常情况下,我们会对API的溶解度、粒径、晶型和制剂的黏度以及均一性等进行控制来保证药品的质量。但是这些物理化学性质都是相对独立的,单独研究无法确定这些性质的组合影响,而体外释放实验(IVRT)能在一定程度上反映各种物理化学性质的组合影响。IVRT不能直接预测药物的体内行为,因为药品的生物利用度和临床表现还与产品所要应用到的上皮细胞的屏障特性,例如表皮和粘膜组织的屏障特性就存在较大的差异。但是其可以检测到可能影响到制剂体内行为的体外变化。引起这些变化的原因主要包括API的物理化学性质、辅料、处方、生产工艺的改变、运输和储存的影响、老化以及其他处方和工艺因素。如果产品出现问题,根据IVRT的结果,研发人员有可能针对性地寻找到解决问题的方法。因此,不管是针对新药批准后工艺或处方的微小变更,还是仿制药研发过程中保证自研与参比质量的一致性,以及监测稳定性试验过程中样品质量的变化,IVRT都有非常重要的意义。
目前,FDA对于IVRT的态度主要有以下几点:1. IVRT只能在特定的批量放大或变更情况下才能证明产品的相似性;2. 不管在NDA还是ANDA申请过程中,IVRT都不是强制的;3. 单独的IVRT不能作为体内生物利用度和生物等效性的替代方法;4. 不使用IVRT比较不同厂家的制剂。
IVRT试验装置
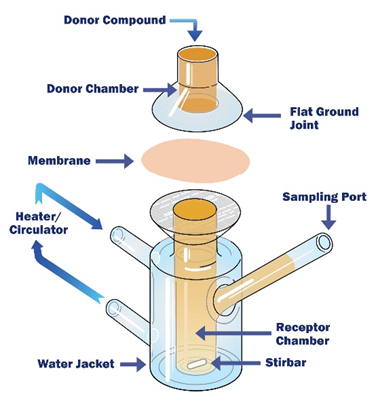
半固体制剂体外药物释放试验方法包括垂直扩散池法、浸没池法、流通池法等,其中垂直扩散池法最为常用,也更容易被审评接受。以垂直扩散池为例,该装置主要包括供给池、隔离薄膜以及带搅拌功能及取样口的接受池,如图1所示。图中隔离薄膜的主要作用是让供给池中的药物与接收池中的介质保持分离。目前,市面上有多种自动化设备可供选择,提供控温、自动取样等功能。
图1:垂直扩散池vertical diffusion cell (VDC)
IVRT试验设计
应为伪无限上药量,整个试验过程中需确保不因上药量低限制药物的释放。FDA推荐的上药量是300mg左右,均匀地铺在薄膜上。也可选择不同厚度,常规有0.5mm、1mm、2mm等。
薄膜仅为保证药物与接收池中的介质保持分离,薄膜必须为惰性且高度渗透的,不与API发生吸附或反应,不阻碍API的扩散。可选择商品化的人工膜如聚砜、混合纤维素酯或聚四氟乙烯等。
筛选介质应考虑API的溶解度及溶液稳定性,保证漏槽条件。优先选用接近生理pH的水性缓冲液。对于难溶性药物可适当加入有机溶剂,如乙醇或表面活性剂。
阴道或直肠制剂选择生理温度37℃,其余选择体表温度32℃
搅拌是为了保证测试过程中接收池中的药物混合均匀,usp<1724>建议常用600rpm,经过筛选的其他转速亦可。
一般建议在6h内取不少于5个点,如30min,1h,2h,4h,6h。
另外,需注意检测IVRT实验过程中的温湿度。FDA推荐的温湿度范围为21℃±2℃;RH50%±20%。
IVRT实验原理及接受标准
伪无限剂量条件下,药物从半固体制剂中释放入漏槽接受池的过程符合Higuchi方程,对于不同的体系,描述药物释放速率的方程略有不同:
若药物均匀地溶解在制剂中,药物释放量vs时间的曲线可由以下方程描述:
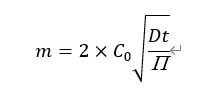
其中,m为每平方厘米药物累积释放量,C0为释放基质中药物的浓度,D为药物通过基质的释放速率,t为时间。
若药物颗粒以混悬状态分散在制剂中,药物释放量vs时间的曲线可以由以下方程描述:
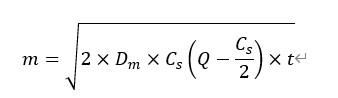
其中,m为每平方厘米药物累积释放量,Dm为药物通过半固体基质的扩散系数,Cs为药物在释放基质中的溶解度,Q为溶解及混悬在基质中的药物总量,t为时间。
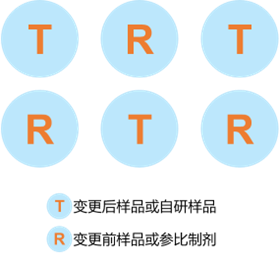
由以上两个方程可以看出:药物的累积释放量与时间的平方根成正比。因此,在实验过程中需要获得药物释放曲线的线性部分,否则IVRT试验不成功。此外,试验设计过程中对同时测定的不同配方的样品的摆放顺序也有一定要求:如下图2所示,待测样品和对照样品应交替放置,以减少试验的系统误差和保证试验的客观性。
图2:IVRT试验待测和对照样品摆放顺序(FDA推荐)
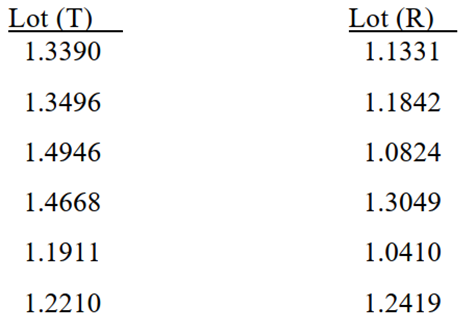
试验结果处理:在预设的时间点取样,并计算药物的累积释放量(μg/cm2),以药物累积释放量为纵坐标,时间的平方根为横坐标作图,所得直线的斜率即为药物释放速率。通常每个配方,平行测定6组。在试验完成后,应利用FDA推荐的统计学方法对得到的数据进行分析,以判定待测样品和参比药品的IVRT是否相似。假设待测组和对照组得到的释放速率数据如下图所示:
将每一个待测样品(T)的释放速率与另外六个对照样品(R)的释放速率相除,这样一共能得到36(6×6)个T/R的比值,将这36个比值从小到大排列,将第8和第29个数值作为T/R比值的90%置信区间的下限和上限,若该置信区间落在75%~133.33%范围内,说明待测样品通过IVRT试验,即IVRT具有相似性,不必再做接下来的试验。若未通过测试,则需再将待测组和对照组再重新各做12次,连同之前测得的6次,共18次,计算得到324(18×18)个T/R的比值,按同样的方法,挑选出第110和215个数值作为T/R比值的90%置信区间的下限和上限,该置信区间应落在75%~133.33%范围内。
方法学验证
人工膜惰性验证:考察所选薄膜对于待测药物的吸附性,选择一个合适药物浓度(与IVRT实验结束时溶液中药物的浓度相当),将人工膜置于所选介质中在32℃条件下培育一段时间(如6h),同时设定无人工膜的对照组,各三份。若最后待测药物的回收率在100%±5%范围内,则说明所选人工膜是惰性的。
与溶出方法的区分力类似,IVRT方法也应具有区分力:灵敏度、特异性、选择性。
灵敏性
:指的是该方法能检测到释放速率随药物浓度的变化而变化,以5%规格药品为例,可以将API含量上下调整到2.5%和7.5%,再进行IVRT试验,考察方法的灵敏性。
特异性
:要求IVRT方法能够准确监测释放速率随药物浓度变化的正相关性,即IVRT释放速率与处方药物浓度成线性,r2≥0.90。
选择性
:指IVRT方法区分对照制剂和改变规格的配方制剂(50%和150%规格)释放速率差异的能力,应证明改变配方制剂规格后,其释放速率与对照制剂相比,在统计学上不等效,即T/R的比值的90%置信区间应落在75%~133.33%范围外。另外,为了进一步支持方法的选择性,还可改变处方的辅料或生产工艺,再进行IVRT研究。
总结
随着药品监管力度的不断加强,现行的审评制度对药企的研发要求也不断提高。在半固体制剂研发中,IVRT用于药物从制剂中释放的速率和程度,是质量研究及稳定性考察中的重要指标,已成为药学研究任务中的“标配”,只有深入理解IVRT的意义及其方法开发和验证方面的细节,才能保证IVRT试验结果的可靠性,夯实药品质量,提高研发效率。
来源:药渡
撰文:woyshu 编辑:丸子
责任编辑:胡静 审核人:何发













评论
加载更多