2022年抗体偶联药物行业研究报告
1 抗体偶联药物:靶向杀伤肿瘤细胞的“生物导弹”
兼具单抗药及细胞毒素优点,ADC 药物临床价值巨大
肿瘤治疗的“生物导弹”,临床价值潜力巨大。抗体偶联药物(Antibody-drug conjugates, ADC)由靶向肿瘤特异性抗原或肿瘤相关抗原的单克隆抗体(Antibody)与不同数目的小分子毒素(Payload)通过连接子(Linker)偶联组成,是近年来肿瘤学发展最快的药物类别之一。由于兼具单抗药物的高靶向性以及细胞毒素在肿瘤组织中高活性的双重优点, ADC 药物可高效杀伤肿瘤细胞,较化疗药物副作用更低,较传统抗体类肿瘤药物具有更好 的疗效,被称为肿瘤治疗领域的“生物导弹”。此外,ADC 药物具备与其他疗法联合的协 同作用并可用于治疗单抗药物疗效不佳的大量潜在患者,展示出巨大的临床治疗价值。
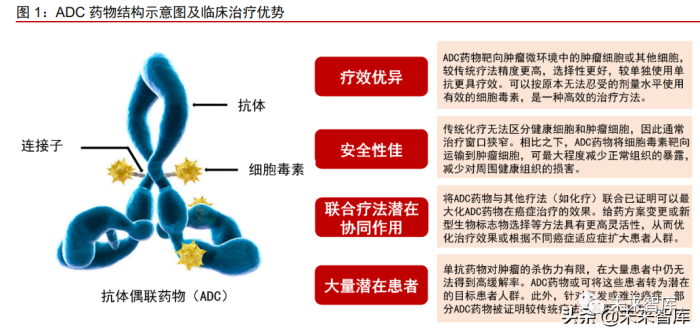
ADC 药物通过抑制肿瘤 DNA 复制或阻滞细胞周期诱导肿瘤细胞死亡。ADC 药物进 入血液循环后,与肿瘤细胞表面靶抗原受体结合,形成 ADC-抗原复合物,被肿瘤细胞内 吞,进而经过溶酶体降解,细胞毒素在胞内释放,结合至 DNA 小沟或微管蛋白,抑制肿 瘤 DNA 复制或阻滞细胞周期,诱导肿瘤细胞死亡。疏水性小分子毒素还可以通过细胞膜扩散,对邻近肿瘤细胞产生杀伤活性,称为“旁观者效应”。对于部分抗体靶标异质性表 达的肿瘤,旁观者效应是 ADC 药物杀伤肿瘤细胞的关键机制。
ADC 药物经历三代变革,技术日臻成熟
第一代 ADC 药物以靶向 CD33 的 Mylotarg 为代表性药物,使用鼠源抗体,免疫原性 较强,易产生人抗鼠抗体;连接子不稳定,毒素在血浆中提前释放导致严重的毒性反应;细胞毒素药物效力不足,不足以杀死肿瘤细胞。
第二代 ADC 药物以靶向 CD30 的 Adcetris 和靶向 HER2 的 Kadcyla 为代表,使用人 鼠嵌合抗体、人源化单抗代替鼠源单抗,采用了毒性更高的细胞毒素和更稳定的连接子, 但药物/抗体比率(DAR)不均一,循环中的未结合细胞毒素的裸抗占比较多,竞争偶联药 物抗原结合位点,降低了疗效;此外,药物过多的与抗体结合易引起抗体聚集、清除速度 加快、非特异性毒性增加等问题。
第三代 ADC 药物得益于定点偶联技术的发展,可将 DAR 值稳定在 2-4 左右,稳定性 和药代动力学得到改善;同时利用可剪切连接子发挥旁观者效应,提升了疗效,降低了毒 性反应,代表药物为靶向 HER2 的 Enhertu。
ADC 药物曲折中发展,市场快速成长
ADC 药物发展曲折中前行,近年来进入爆发阶段。早在 1913 年,德国诺贝尔奖得主 Paul Ehrlich 就首次提出了 ADC 药物的设想。1958 年 Mathe 首次将抗鼠免疫球蛋白与 MTX 偶联用于治疗白血病,但由于免疫原性等问题,实验未获成功。直到 1975 年,杂交 瘤技术生产单克隆抗体的发展正式拉开了 ADC 药物研发的序幕。利妥昔单抗和曲妥珠单 抗的上市为 ADC 药物的面市进一步奠定了基础。2000 年,首款 ADC 药物辉瑞的 Mylotarg 获 FDA 批准上市用于治疗急性髓系白血病(AML),但由于连接子不稳定、细胞毒素提前 释放引起严重毒性反应,Mylotarg 于 2010 年撤市。随后辉瑞调整剂量,并补充更多数据 后,该款药物的收益-风险比终于获得认可,2017 年 Mylotarg 重新上市。从 Mylotarg 首 次上市到再次上市的 17 年间,一共仅有 3 款 ADC 药物获批上市,随后在生物技术的不断 突破下,ADC 药物发展逐步成熟,上市品种快速增加。
全球共有 14 款 ADC 药物获批上市,国内已获批 4 款。截至目前,全球已有 14 款 ADC 药物获批上市,7 款用于血液肿瘤,7 款用于治疗实体瘤,靶点涉及 CD33、CD30、CD22、 CD79b、HER2、Nectin-4、Trop-2、BCMA、EGFR、CD19 和 TF。其中,4 款 ADC 药 物在中国上市。荣昌生物自主研发的维迪西妥单抗于 2021 年 6 月 9 日在国内获批上市, 成为首个获批的国产 ADC 新药。此外,Adcetris 和 Kadcyla 于 2020 年获 NMPA 批准在 中国上市, Besponsa 于 2021 年 12 月在中国获批上市。越来越多的 ADC 药物获批激发 了药企对于 ADC 的研发热情,随着临床进展的推进,未来将有更多 ADC 药物及适应症推 进到临床后期进而获批上市。
全球 ADC 药物市场规模快速增长。已经上市的 ADC 药物在全球市场各个地区均展现 出良好的销量增长态势。
美国:首款 ADC 药物在美国上市,目前在上市药物数量及市场规模均遥遥领先 于其他地区。自 2011 年以来共有 11 款 ADC 药物获批上市,2016-2020 年,美 国 ADC 药物市场规模从 3.29 亿美元增长至 14.70 亿美元,CAGR 高达 45%, 是全球增速最快的地区。
欧洲:目前共有 7 款 ADC 药物获批,2016-2020 年,欧洲地区 ADC 药物市场规 模从 3.59 亿美元增长至 5.81 亿美元,CAGR 为 13%。
日本:自 2014 年以来共有 4 款 ADC 药物获批,2016-2020 年,日本 ADC 药物 市场规模从 1.03 亿美元增长至 2.22 亿美元,CAGR 为 21%。
中国:全球上市较早的 Adcetris 和 Kadcyla 2020 年在中国获批上市,2020 年两 款药物合计销售额为 57 万美元。2021 年 6 月,首款国产创新 ADC 药物-荣昌生 物的 RC48(爱地希)上市,目前共有 3 款 ADC 药物在中国上市销售。中国刚 刚迎来 ADC 药物的研发热潮,我们预计市场将在未来 5 年内快速成长。(报告来源:未来智库)
2 全球 ADC 研发热情高涨,差异化竞争是关键
全球 ADC 热门靶点集中,适应症以实体瘤为主
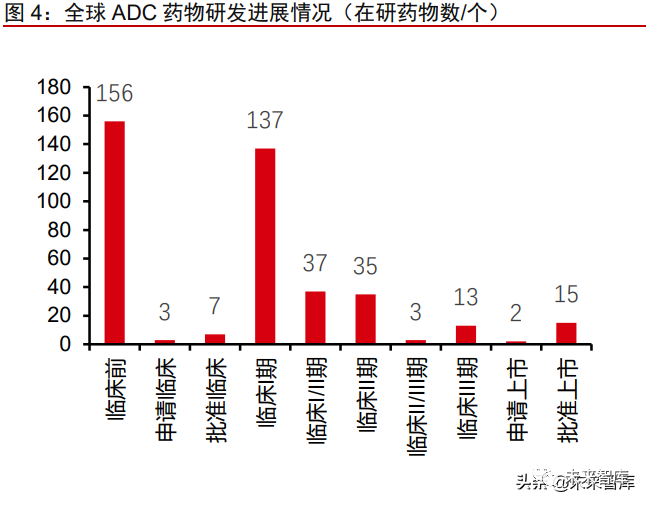
全球共有 408 款 ADC 药物在研,大多数处于临床早期。近年来,ADC 药物在全球掀 起研发热潮,成为众多新药研发企业重点布局领域。根据 Insight 数据库统计,截至 2021 年 12 月 29 日,全球共有 408 个 ADC 药物处于不同研发阶段。目前大部分 ADC 药物处 于临床早期,仅 15 款产品已上市(占比 4%,其中一款为印度上市的 Kadcyla 仿制药), 13 个候选药物进入临床 III 期阶段(占比 3%),而处于临床 I 期、临床前阶段的药物高达 137、156 款,分别占比约三分之一。
全球 ADC 药物研究靶点集中在经验证的成熟靶点,热门靶点药物竞争日趋激烈。全 球已知靶点的 310 个 ADC 药物(408 个药物中,98 个靶点尚未披露),较为热门的靶点 包括 HER2、CLDN-18.2、CD19、FRα、CD33、ROR1、CD70、CD276、IL3RA、DLK1 等,大多为已有药物获批上市、经验证的成熟靶点。其中在研药物数量最多的是 HER2, 共有 40 个,CLDN-18.2 排名第二,共有 14 个。此外,103 个 ADC 药物布局靶点没有同 类产品在研(占比 25%)。
全球 ADC 药物适应症以实体瘤为主,并向眼病、自身免疫等领域延伸。从全球在研 ADC 药物适应症布局情况看,肿瘤占绝大多数。与已上市的药物的适应症布局情况不同(14 款上市产品中,7 款用于血液瘤治疗,7 款实体瘤),在研 ADC 药物更多往实体瘤延伸(占 比高达 64%),主要集中在乳腺癌、肺癌、胃癌等患病人群较多的癌种,血液瘤占比 18%。其他适应症包括类风湿性关节炎、AL 淀粉样变性、金黄色葡萄球菌血症、移植物抗宿主 病以及年龄相关黄斑变性等眼病。
国内 ADC 研发仍处于起步阶段,后期管线以引进为主
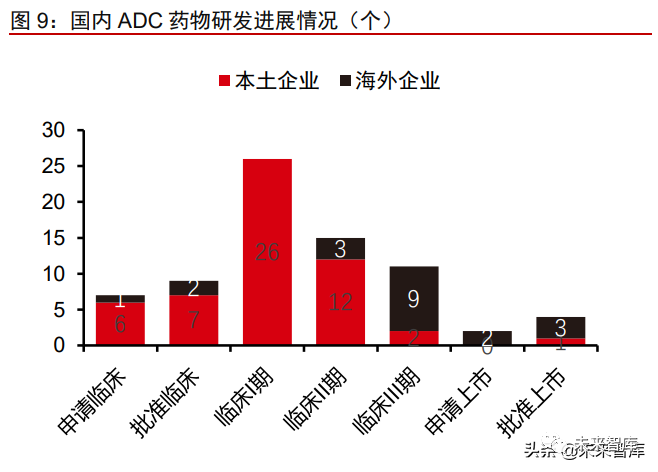
国内药企 ADC 药物研发仍处于起步阶段,后期管线产品以外企及联合开发为主。根 据 Insight 数据库统计,截至 2021 年 12 月 29 日,国内共有 74 个 ADC 药物处于不同研 发阶段。其中临床 I 期占比最高(为 35%),这部分药物以国内企业自主研发为主,代表 企业包括荣昌生物、科伦药业、恒瑞医药、多禧生物等。处于临床后期(III 期及以上)的 产品共有 17 款,其中本土企业申报的产品仅 3 款,该部分药物主要由海外药企开发,多 是海外已经上市或进入临床后期的产品。除荣昌生物、东曜药业、百奥泰(已终止)的 3 款药物为自主研发外,其余均由外企研发。国内药企为实现 ADC 药物的快速上市,积极 引进海外临床后期的 ADC 产品,包括云顶新耀、华东医药和浙江医药等。
国内 ADC 药物研究靶点扎堆 HER2 和 TROP2,血液瘤相关靶点布局相对较少。国 内已知靶点的 69 个 ADC 药物(74 个药物中,5 个靶点尚未披露),较为热门的靶点包括 HER2、TROP2、CLDN-18.2、MET、NECTIN4、CD79B、TNFRSF8、CD20、MSLN 和 EGFR。相比全球热门靶点,CD22、BCMA 等靶向血液瘤的靶点布局明显减少。其中 在研药物数量最多的是 HER2,共有 22 个,TROP2 排名第二,共有 7 个。国内约三分之 一的 ADC 药物靶向 HER2,目前除 2 款已获批上市,4 款处于临床 III 期外,尚有 9 款处 于临床 I 期。随着越来越多 ADC 药物布局靶向 HER2,预计该靶点的 ADC 药物将面临较 为激烈的竞争格局。
国内 ADC 药物适应症以实体瘤为主,尚未有本土药企布局肿瘤以外领域。从国内在 研 ADC 药物适应症布局情况看,实体瘤占绝大多数(70%),血液瘤次之(15%),与全 球适应症布局基本相似。而其他适应症仅有 Kodiak Sciences 的 KSI-301 布局糖尿病黄斑 水肿等眼科疾病,尚未有本土药企涉足肿瘤以外的领域。
ADC 药物市场潜力巨大,全球掀起合作/并购热潮
最早上市 10 款 ADC 药物市场潜力巨大,预计 2026 年全球 ADC 药物市场规模有望超过 400 亿美元。根据发表在 Nature Reviews 的《The oncology market for antibody-drug conjugate 》(Carolina do Pazo 等)一文,最早上市的 10 款 ADC 药物市场规模预计到 2026 年将超过 164 亿美元。其中 trastuzumab deruxtecan(Enhertu,阿斯利康/第一三共) 已获批多个乳腺癌(含 HER2+、HR+/HER2-及三阴性)适应症,并且用药时间较长,将 以 62 亿美元的销售额成为最畅销的 ADC 药物。Enfortumab vedotin(Padcev,Seagen/ 安斯泰来)获批治疗经治的转移性尿路上皮癌,未来有望适应症拓展至患者人群更多的早 期阶段,2026 年全球销售额有望达到 35 亿美元,位居第二位。在全球 ADC 药物研发热 潮下,随着未来更多其他 ADC 药物获批以及适应症的不断拓展,销售额有望快速增长。假设到 2026 年,最早上市的 10 款 ADC 药物占据 40%市场份额,则全球销售额有望达到 410 亿美元。
经我们测算,HER2 ADC 药物 2025 年国内市场空间超 30 亿元。HER2 靶点的扎堆 也反映该靶点 ADC 药物较大的市场空间。国内靶向 HER2 的 ADC 药物适应症集中在晚期 乳腺癌、胃癌及尿路上皮癌,代表性药物包括罗氏/ImmunoGen 的恩美曲妥珠单抗,荣昌 生物的维迪西妥单抗。我们以晚期乳腺癌、胃癌及尿路上皮癌这三个主要的适应症测算国 内 HER2 ADC 药物的市场空间。根据我们测算,到 2025 年国内 HER2 ADC 药物用于治 疗晚期乳腺癌、胃癌及尿路上皮癌的市场规模分别为 17.17 亿、13.84 亿、0.36 亿,合计 31.37 亿元。
国内药企 ADC 药物研发仍处于起步阶段,荣昌生物进度国内领先。相比美国,国内 ADC 药物起步较晚,首款 ADC 上市时间间隔 21 年。进展最快的荣昌生物维迪西妥单抗 于 2021 年 6 月获批上市,成为国内首款上市的国产 ADC 药物,此外进度较快有正在申报 上市的云顶新耀(与 Immunodedics 合作),处于临床 III 期的东曜药业(自主研发)、齐鲁 制药(与 Sesen Bio 合作)、浙江医药(自主研发)、华东医药(与 ImmunoGen 合作)、 缔脉生物(与 Kodiak Sciences 合作)。恒瑞医药布局的 ADC 药物尽管仍处于研发早期, 管线数量国内最丰富(1 款 II 期、6 款 I 期,靶点涉及 HER2、c-MET 等)。
全球掀起 ADC 药物合作/并购热潮,争取快速布局 ADC 药物赛道。国内外众多企业 看到 ADC 药物巨大的临床潜力以及不断扩展的适应症,为了瓜分 ADC 药物广阔市场空间 的份额,在自主研发的同时,通过战略合作,并购等方式快速布局 ADC 药物赛道。其中 较为著名的是阿斯利康接连与第一三共签订三款 ADC 药物(靶向 HER2、TROP-2、HER3) 的合作,以及吉利德以约 210 亿美元收购 Immunomedics,获得了其一款靶向 TROP2 的商业化 ADC 产品 Trodelvy 全球首个上市的靶向 TROP2 的抗体偶联物。(报告来源:未来智库)
3 ADC 研发壁垒高,三大元件不断优化
抗体:高特异性、内吞作用,决定 ADC 疗效的关键因素
不同的嵌合抗体的免疫原性程度不同,降低鼠源性有利于解决异种蛋白的排斥问题。早期 ADC 药物采用鼠源抗体,易引起急性超敏反应或产生人抗鼠中和性抗药抗体,第二、 三代 ADC 药物中,人源化抗体广泛使用,极大的规避了上述问题。
已上市 ADC 药物抗体种类以 IgG1 为主。从抗体种类来看,目前所有在研 ADC 药物 均采用免疫球蛋白 G(IgG),这种生物分子具有多个天然结合位点,并可被进一步修饰产 生新的活性位点,此外,IgG 对于靶抗原具有高亲和力,在血液循环中半衰期较长,因此 是 ADC 药物抗体部分的理想选择。IgG 共有四种亚型(IgG1、IgG2、IgG3 和 IgG4),已 上市 ADC 药物中,除辉瑞的 Mylotarg 和 Besponsa 采用 IgG4 抗体,其余均选择 IgG1 作 为抗体。
IgG1 具有较强 ADCC、CDC 效应及更长半衰期。IgG1 和 IgG3 相较于 IgG2 和 IgG4 具有更强的抗体依赖的细胞毒效应(ADCC)和补体依赖的细胞毒效应(CDC),可与细 胞毒素形成协同作用杀伤肿瘤细胞,然而 IgG3 分子循环半衰期较短,血浆清除速度过快, 因此不是 ADC 抗体的最佳选择。也有部分研究者认为 ADCC 作用加上小分子毒性药物将 导致 ADC 药物毒性过强,但目前尚无定论。此外,IgG2 和 IgG4 铰链区二硫键较难还原, ADC 生产难度较大,并且 IgG4 抗体易发生 Fab 臂交换,形成新的杂合 IgG4,低 pH 条件 下稳定性不及 IgG1。但对 IgG4 的重链改造可有效解决这一问题,因此对不同抗体亚型的 修饰以增强稳定性或抗体特异性可能是未来研发的新方向。
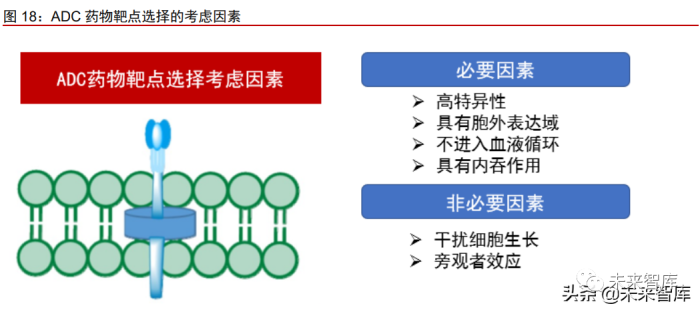
理想的抗原应具备以下几个特征:(1)抗原具有高特异性。即在肿瘤细胞高表达而在 正常细胞低表达,以减少 ADC 药物的脱靶效应;(2)目标抗原应具有胞外表达域,以便 循环中的抗体结合并进入目标细胞;(3)抗原不分泌至胞外进入血液循环,以避免抗体在 循环中与游离抗原结合,导致全身性毒性;(4)抗原具有内吞作用,使其与抗体结合后, ADC 药物能被转运到细胞内,介导细胞毒素发挥杀伤作用。
抗原选择的“加分项”而非必要因素包括:(1)干扰细胞生长。ADC 药物对细胞生 长的抑制作用主要通过内化而非抑制细胞生长。然而结合可参与细胞分裂通路的抗原(譬 如 CD30、CD70)可增长 ADC 药物的疗效;(2)旁观者效应。肿瘤细胞具有一定的异质 性,抗原-抗体结合诱导的 ADC 药物内吞仅对部分肿瘤细胞有效。部分抗原表达的肿瘤细 胞被杀伤后会释放活性氧自由基、细胞毒性代谢物及 ADC 药物到周围的肿瘤细胞。
理想的抗体应具备以下几个特征:(1)高特异性及低抗原免疫原性是 ADC 药物抗体 的主要特征。可避免抗体非特异性结合到其他抗原造成对正常细胞的损伤,或在抵达目标 肿瘤细胞前的消耗;(2)抗体的高亲和力。抗体与抗原较高的亲和力可促进 ADC 药物的 高效内吞;(3)受体介导的抗体内吞。《Case Study: An antibody-drug conjugate targetingMUC16 for ovarian cancer.》(Leipold D et.al)一文的研究表明针对同一抗原的不同抗体具有不 同的程度的内吞作用,快速的内吞作用可减少 ADC 的脱靶效应,从而提高药物疗效及安 全性。此外,内吞的途径也十分重要,假设 ADC 药物内吞后被运输到高尔基体或内质网 (非水解环境)而不是溶酶体(可水解环境),则将大大影响 ADC 药物细胞毒素的释放。
抗体选择的“加分项”而非必要因素为抗体本身的治疗作用。抗体的治疗作用通常是 通过免疫反应介导,包括抗体介导的细胞毒性作用(ADCC)、抗体介导的细胞吞噬作用 (ADCP)、补体介导的细胞毒性作用(CDC)以及细胞因子激活通路的调节,这些治疗作 用可提升 ADC 药物对目标细胞的杀伤作用。
连接子:精准切割、高均一性,决定 ADC 安全性的关键因素
连接子是决定 ADC 药物系统毒性及临床疗效的关键组分。连接子的作用主要体现在 两个方面:(1)确保当 ADC 药物仍在血液循环中时,细胞毒素紧紧与单抗连接。在血液 中不稳定的连接子会导致细胞毒素提前释放,致使严重的全身性毒副作用,并且降低了抵 达肿瘤组织的 ADC 药物数量;(2)确保当 ADC 进入肿瘤细胞后,细胞毒素可有效释放。由此可见,连接子是决定 ADC 药物系统毒性及临床疗效的关键组分。
连接子可大致分为两类:可切割连接子(cleavable linker)和不可切割连接子 (non-cleavable linker)。二者在二三代 ADC 药物中均有应用。可切割连接子利用肿瘤细 胞(低 pH,还原环境)和循环内环境(如溶酶体的蛋白水解酶)差异断裂并释放细胞毒 素。
可切割连接子:可分为三种类型:(1)酸依赖性腙键(代表性药物如 Mylotarg);(2) 氧化还原依赖的二硫键;(3)可酶切肽键。然而,在临床上这些可剪切连接子在血液循环 中都有不同程度释放,导致系统性毒性,这也是 Mylotarg 因毒性较高撤出市场的主要原因 之一。
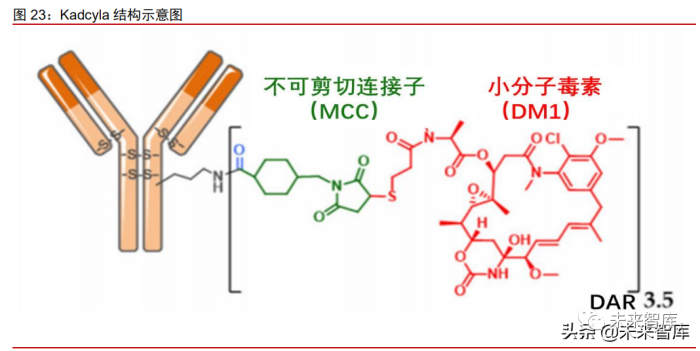
不可切割的连接子:(譬如硫醚键--代表性药物 Kadcyla)的优势在于血液环境中更加 稳定,仅在溶酶体中切割释放细胞毒,安全性明显更好。然而不可切割连接子通常会残留在细胞毒素上,影响其杀伤活性及细胞穿透性。因而对周围的异质性肿瘤细胞杀伤作用有 限,不能充分发挥旁观者效应。
提高产品均一性,定点偶联有效拓宽治疗窗
传统随机偶联方式产生的 ADC 为异质混合物,均一性和稳定性差。ADC 利用连接子 将单抗与细胞毒素连接,传统 ADC 药物多采用随机偶联方式,利用赖氨酸的氨基或打开 链间二硫键获得的半胱氨酸的巯基进行偶联,难以控制偶联位置和数量。因此传统的 ADC 药物是高度异质混合物,包含了不一样的 DAR 和不一样的偶联位点,稳定性差,易发生 聚集,且其中的细胞毒素容易脱落而产生非治疗性毒副作用均一性较差,稳定性低,影响 药效与治疗窗口。
赖氨酸偶联:抗体表面的赖氨酸能和连接子上的琥珀酰亚胺(NHS)酯反应,从而构 建 ADC。IgG 分子具有超过 80 个赖氨酸残基,其中溶剂可及性高的位点超过 20 个,这些 氨基酸均可被酰化偶联导致异质混合物的产生,因此采用赖氨酸偶联得到的 ADC 药物异 质性较强。使用该偶联法的代表性 ADC 药物有 Kadcyla、Mylotarg 及 Besponsa。
二硫键还原半胱氨酸偶联:通过抗体上的半胱氨酸和连接子上的马来酰亚胺发生反应 构建 ADC。IgG 序列内的半胱氨酸残基较赖氨酸数量少很多,一个 IgG 含有 16 对二硫键, 其中 12 对为链内,4 对为链间。4 对链间二硫键在还原剂作用下可还原出 8 个具有亲核性 能的巯基,是偶联的潜在位点。相较赖氨酸偶联,该偶联方式显著降低 ADC 的异质性, 得到均一性更高的 ADC。然而这种偶联方式的二硫键可完全或部分还原,导致产生的 ADC 每个抗体最多有 8 个有效载荷,仍会产生不均一的 ADC 药物。该法是目前使用最多的偶 联方式,譬如 Adcetris、Trodelvy 和 Enhertu。
近年来定点偶联技术的出现实现了抗体和小分子毒素的定点定量偶联,均一稳定,具 有更好的活性和药动学特性,有效拓宽了治疗窗,极大推动了 ADC 领域的发展。定点偶 联技术主要分为五类:Thio-Mab 技术、引入非天然氨基酸、糖基偶联、氨基定点偶联和 酶促多肽偶联。
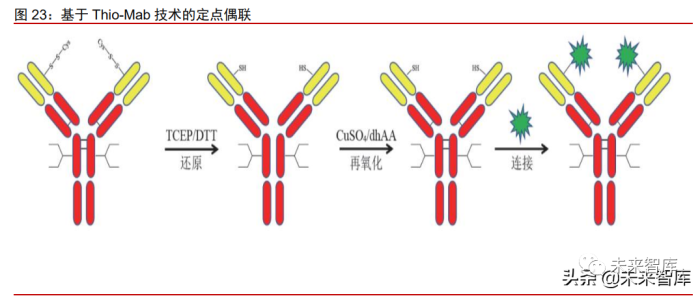
Thio-Mab 技术:Thio-Mab 技术由 Genetech 首先开发,在抗体 Fab 表面筛选出 反应性半胱氨酸的突变位点(曲妥珠单抗的 LC-V110 和 HC-A114)进行半胱氨 酸突变,利用三(2-羧乙基)膦(TCEP)或二硫苏糖醇( DTT)只打开抗体链 间二硫键,并使突变的半胱氨酸的巯基处于自由态,再应用 CuSO4 或脱氢抗坏 血酸( dhAA)将链间二硫键重新连接,最后利用游离巯基与药物连接子反应, 实现抗体药物定点偶联。
引入非天然氨基酸:该技术通过引入可特异性识别非天然氨基酸的 tRNA 和氨酰 tRNA 合成酶,与对应的非天然氨基酸结合形成氨酰 tRNA,再通过其反密码子与 mRNA 上的密码子互补,将非天然氨基酸整合到多肽链中。引入的非天然氨基酸 上的酮基或叠氮基官能团可与连接子-毒素定向偶联。形成的化学键非常稳定,在 血液中不易解离。Ambrx开发的EuCODE平台和Sutro Biopharma开发的Xpress CF+平台分别利用引入非天然氨基酸的酮基和叠氮苯丙氨酸残基开发了 ARX788 和 Stro-001、Stro002。
糖基偶联:Synaffix 的 GlycoConnect 技术利用糖苷内切酶使天然抗体上的糖基 暴露出 N-乙酰氨基葡萄糖,进而将经叠氮修饰后的 N-乙酰半乳糖胺利用糖基转 移酶连接到抗体的 N-乙酰氨基葡萄糖上,被叠氮修饰后的抗体进一步与双环酮 修饰的连接子-毒素发生点击化学反应,形成稳定的共价键,得到定点偶连的 ADC。2019 年 4 月,上海美雅珂以 1.25 亿美元引入 Synaffix 的 GlycoConnect 和 HydraSpace ADC 技术,用于开发 ADC 药物。
特定赖氨酸的氨基定点偶联:由于抗体上的赖氨酸数目众多,利用赖氨酸偶联将 导致位置和数量高度不均一。氨基定点偶联策略利用赖氨酸在特定 pH 下与亲核 试剂的反应速率差异,选择合适的连接子,可选择性与特定赖氨酸残基偶联。四 川科伦的 A166 是全球首个基于此技术开发的 ADC 药物,用于治疗 HER2 阳性乳腺 癌。
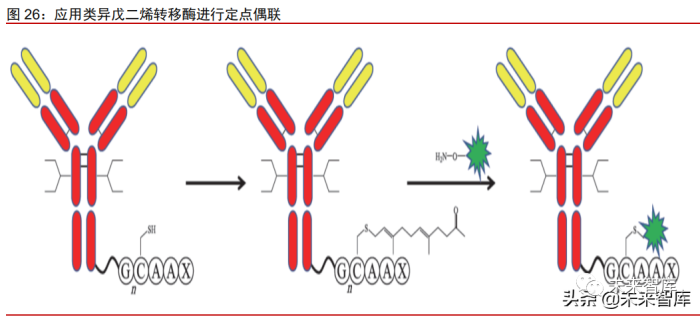
酶促多肽偶联:利用基因工程技术使抗体中产生能被某些酶识别的相关氨基酸序 列,再利用酶对底物的特异性将其中的特定氨基酸残基进行改造,从而实现定点 偶联。主要包括 SMARTag 技术、ConjuAll 技术等,分别利用甲酰甘氨酸合成酶 (FGE)和类异戊二烯转移酶。由 LegoChem Biosciences 与复星医药合作开发 的 FS-1502(LCB14-0110)采用的就是 LegoChem 公司的 ConjuAll 技术。
由于定点偶联对 ADC 药物的活性和药物动力学特性都有至关重要的作用,定点偶联 仍是以后 ADC 发展的方向,推动 ADC 药物向这高均一性、高稳定性和高药效性发展。目 前已上市的 ADC 药物大多采用随机偶联方式。随着定点偶联技术的发展,ADC 药物将展 现出更大的临床治疗价值。
细胞毒素:高毒性、高稳定性,决定 ADC 活性的关键因素
细胞毒素是决定 ADC 药物杀伤力大小的关键因子。理想的细胞毒素应该具有以下几 个特性:
具有高毒性:通常中度毒性的细胞毒素分子杀死单个细胞需要的数量大于 10 6, 而每个肿瘤细胞表面大约存在 10 5 个抗体能结合的抗原。并且,从 ADC 药物的 细胞毒素通过抗体抗原结合内吞进入细胞、到在细胞内释放出细胞毒素分子的过 程中,实际能够进入到细胞的细胞毒素分子数很低(被内化的抗体不到注射剂量 的 0.1%)。因此 ADC 药物携带的细胞毒素首先有具有很强的毒性(IC50 应小于 10nM)。
低免疫原性:高毒性的细胞毒素可以在植物、动物或微生物中提取,确保细胞毒 素在人体中的免疫原性小到可以忽略不计。
高稳定性:细胞毒素应该在药物制备和储存时维持稳定;在血液循环中能保持稳 定性,因为细胞毒素会长时间在血液中循环,要避免其在血液中释放或分解;在 细胞质和溶酶体中保持稳定,不受低 pH 条件影响而分解。
具有可修饰的官能团:细胞毒素分子要有可修饰的官能团或可引入官能团的位点, 能进一步与单克隆抗体相连。
ADC 药物所用细胞毒素可分为三类,微管蛋白抑制剂最为常见。早期 ADC 药物开发 选用了临床上常用的化疗毒性药物,包括多柔比星、甲氨蝶呤、丝裂霉素、氟尿嘧啶及长 春碱等,但与抗体连接后,在低浓度下杀伤肿瘤细胞的效果太弱,因此不能作为 ADC 药 物的细胞毒素。目前临床试验或已上市 ADC 药物所用的细胞毒素约 6-8 个,主要是天然 来源,按作用方式可大致分为三类:(1)微管蛋白抑制剂(奥利斯他汀类 Auristatins 和美 登素生物碱类 DMs);(2)DNA 合成抑制剂(卡奇霉素 calicheamicin、多米卡星 duocarmycin,及吡咯并苯二氮䓬类 pyrrolobenzodiazepine,PBDs 等);(3)拓扑异构酶 抑制剂和 RNA 聚合酶 II 抑制剂(喜树碱衍生物和α- 鹅膏蕈碱)。其中最常见的是微管蛋 白抑制剂,占临床开发中的 ADC 药物的一半以上。
微管蛋白抑制剂:多拉司他汀 10(dolastatin 10) 是 1987 年报道的海洋来源的毒性 多肽分子, 能够抑制有丝分裂,但由于毒性太大而不能单独作为药物使用。奥利斯他汀类 是多拉司他汀 10 的衍生物,具有较大的细胞毒性;其中单甲基 auristatin E(MMAE) 和单 甲基 auristatin F(MMAF) 具有更好的水溶性、稳定性及易于引入连接基等特性,能够引起 细胞周期 G2/M 期的阻滞,是常用 ADCs 细胞毒分子。(使用 auristatin 的代表性 ADC 药物:Adcetris、Polivy、Padcev、Blenrep);DMs 是天然化合物美登素的硫醇化合物, 与长春碱对微管蛋白的作用位点相同,但具有更强的毒性。其中,DM1 和 DM4 是 ADCs 中常用细胞毒分子,同样能引起细胞周期 G2/M 期的阻滞(使用 DMs 的代表性 ADC 药 物:Kadcyla)。
DNA 合成抑制剂:卡奇霉素是从棘孢小单胞菌中分离得到的烯二炔化合物,能够与 DNA 序列 TCCTAGGA 的小沟结合,引起双链断裂;多米卡星是从链霉菌属细菌中分离出 来的抗菌素,其细胞毒性能达到皮摩尔级别,能够烷基化 DNA 中富含 AT 碱基的小沟,引 起 DNA 断裂;PBDs 同样是从链霉菌属细菌中分离得到的抗肿瘤抗菌素,能够结合到 DNA 小沟上,识别 5’-嘌呤-G-嘌呤-3’-序列,与鸟嘌呤碱基的 C2 位氨基共价结合,具有较强的 毒性。
拓扑异构酶抑制剂和 RNA 聚合酶 II 抑制剂:喜树碱衍生物 SN-38 和 DX-8951f 是目 前用于 ADCs 的 2 种细胞毒分子,作用于拓扑异构酶Ⅱ。SN-38 是抗肿瘤药物伊立替康(半 合成水溶性喜树碱类衍生物)的活性代谢物,毒性比伊立替康强 3 个数量级,但溶解性较差, 不能直接作为药物使用,然而可将其作为细胞毒分子与单抗相连形成水溶性的 ADCs 使用。DX-8951f 是依沙替康的甲磺酸盐,是一种水溶性抗肿瘤药物,抗肿瘤活性强于其他喜树 碱衍生物,并且能有效阻止 P-糖蛋白(P-gp)介导的多药耐药,也被用于 ADCs 研究。(使 用喜树碱衍生物的代表性 ADC 药物:Enhertu 和 Trodelvy);α-鹅膏蕈碱是双环八肽毒素, 对 RNA 聚合酶Ⅱ具有较高的选择性,目前用其作为细胞毒分子的 ADCs 都处在临床前研 究阶段。
目前普遍认为,单个抗体携带 2~4 个细胞毒分子(即 DARs 为 2~4)的 ADCs 具有较 佳的抗肿瘤活性且具有较低的不良反应。
ADC 药物技术壁垒高,催生 CDMO 业务旺盛需求
研发生产面临挑战,CDMO 行业赋能 ADC 药物开发
ADC 药物研发生产技术复杂,对药企相关能力要求较高。ADC 药物极大的治疗潜力 使其成为国内外众多创新药企业加码布局、研发的热门方向。ADC 药物的设计研发不是抗 体、连接子、细胞毒素三部分的简单结合,需要选择合适的组合使整体疗效和安全性达到 最优。生产工艺同样复杂,包含了单抗、原液、制剂等多个生产环节,尤其是生物偶联技 术,门槛高,难度大。这些因素对 ADC 药物原研企业的研发生产及商业化能力提出巨大 挑战。
CDMO 企业赋能新兴 Biotech 企业进行 ADC 药物创新研发。国内 ADC 研发管线中 新兴 Biotech 公司占比大,绝大多数原研团队无法同时兼具生物制剂、小分子、偶联技术 的开发生产能力。研发、生产技术的高壁垒促使大多数药企寻找合同研发生产组织(CDMO) 机构赋能创新。CDMO 企业可提供包括抗体、连接子、细胞毒素以及偶联技术的开发服务, 以及后期临床阶段所需药物的生产服务。据了解,全球大多数 ADC 药物以外包为开发形 式,借助 CDMO 企业更快进入临床阶段以及市场。海外公司为了控制研发成本也选择价 格低廉的国内 CDMO 企业进行合作。
同类型靶点扎堆布局加剧 ADC 药物竞争,ADC 药物的 CDMO 市场需求巨大。伴随 国内外 ADC 领域进入快速发展阶段,尤其是同类型靶点的竞争加剧,高效的临床推进和 差异化的适应症开发策略成为 ADC 药物布局药企未来取得市场竞争优势的重心,因此选 择合适的 CDMO 伙伴合作也是这些药企研发生产的重要策略。CMDO 市场存在大量的未 满足需求,仅少数几家本土企业布局 ADC 赛道 CDMO 服务领域,如药明生物、东曜药业、 迈百瑞生物、凯莱英医药和臻皓生物。此外,药品上市许可持有人制度(MAH 制度)等 新政的实施推动 CDMO 公司更快发展。在市场需求和政策扶持的共同作用下,国内 CDMO 行业加速发展,未来 ADC 药物 CDMO 市场空间将非常广阔。
响应市场需求,CDMO 企业打造“端到端”一体化平台
多家 CDMO 企业响应市场需求,打造 ADC 产品“端到端”一体化服务平台。过去, ADC 原研企业需要寻找多家 CDMO 企业分别就单抗、细胞毒素、连接子和偶联等不同领 域展开合作,且不同 CDMO 可能分布在不同国家和地区,一方面导致研发生产效率低下, 另一方面为生产和供应链管理带来诸多困难。如今,ADC 药物研发和商业化浪潮已至,对 产品开发速度要求也不断升高。多家 CDMO 企业响应市场需求,打造 ADC 产品“端到端” 一体化服务平台,行业内呈现扩张和整合的态势。近期,ADC 药物 CDMO 领域有两项重 大合作:药明生物和合全药业成立药明合联,聚焦偶联药物的 CDMO 服务;东曜药业与 博瑞医药强强联合,优势互补。2020 年年底,迈百瑞生物也将 CDMO 服务产业链从研发 阶段扩展至商业化生产。
药明合联成立,打造全球领先一体化 CDMO 服务平台。药明生物是全球大分子领域 CDMO 龙头企业,早在 2013 年就开始布局 ADC 市场。目前在 ADC 领域,药明生物共开 展 40 多个项目,其中还包括 1 个即将进入商业化的 III 期临床项目。药明康德子公司合全 药业是聚焦于小分子药物的 CDMO 企业,掌握了先进的高活性毒素分子和连接子的工艺 技术,为小分子的评估提供极大灵活性。2021 年 5 月,药明生物与合全药业成立合资公 司药明合联,该公司将专注于 ADC 和其他生物偶联药物的 CDMO 服务,建立起 ADC 药 物研发生产端对端的一站式服务平台,提供 ADC 从提出概念到商业化生产所需要的一整套服务。药明生物和合全药业曾多次紧密合作,为全球 ADC 研发团队提供 CDMO 服务。如今两个公司强强联手,成立合资企业,将更好地整合双方优势资源,推动 ADC 药物更 快上市。
东曜药业 CDMO 业务持续扩大,与博瑞医药强强联合。东曜药业专注于肿瘤创新药 的研发与商业化,也是国内首批布局 ADC 赛道的公司。利用自身平台及商业化生产优势, 东曜药业开启了 CMO/CDMO 的业务模式。多年耕耘,公司搭建起 ADC 药物的全产业链 平台,从药物开发到产业化形成一整套服务体系。生产方面,东曜药业拥有完整的 ADC 分析、偶联工艺及放大技术,能够在同一个生产车间集中完成单抗、ADC 原液和制剂的关 键生产环节,大大降低了风险管控难度。7 月 19 日,东曜药业与博瑞医药宣布达成战略合 作协议,共同开展 ADC 领域的 CDMO 服务。博瑞医药在复杂小分子的研发和注册方面有 丰富的经验,未来将负责提供连接子和小分子毒素等中间体,与东曜药业实现资源互补。
迈百瑞是国内 ADC 领域的 CDMO 头部企业,拥有先进的 ADC 技术平台。迈百瑞总 裁李新芳博士曾在全球率先从事 ADC 药物研发的公司 ImmunoGen 工作十三年,深度参 与多个 ADC 研发项目。李新芳博士自 2018 年加入迈百瑞后,加速公司成为 ADC 领域 CDMO 赛道上的领跑者。2019 年,迈百瑞荣获 Frost&Sullivan 颁发的“ADC 药物 CMO 全球成长卓越领导奖”。2020 年年底,公司 ADC 专用商业化制剂线正式投产,借此,迈 百瑞已具备 ADC 药物从早期开发到商业化生产的全链条服务能力:包括 ADC 所需的抗体、 连接子和小分子毒素生产、ADC 偶联和制剂生产等。目前在国内借助于 CDMO 获得临床 试验批件的 ADC 新药中,迈百瑞为半数以上的项目提供了服务,印证了公司在国内 ADC 领域 CDMO 行业的服务品质和头牌地位。
4 海外 ADC 上市药物疗效优异,标准疗法不断被刷新
Adcetris:为 CD30 阳性淋巴瘤患者带来新希望
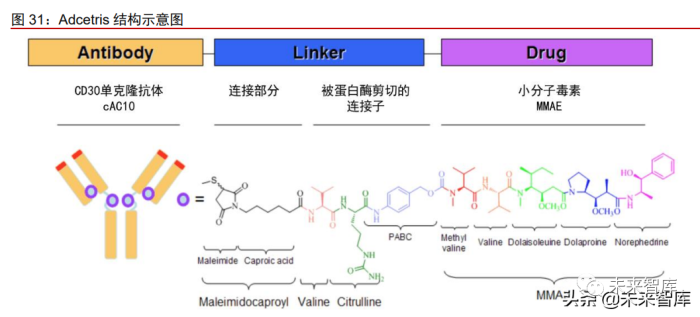
Adcetris(brentuximab vedotin)是全球第一款获批上市的第二代 ADC 药物。 Adcetris 由 Seagen 公司研制,从 2011 年上市至今已围绕治疗经典霍奇金淋巴瘤(cHL) 和系统性间变性大细胞淋巴瘤(sALCL)获批 6 个适应症。2009 年武田与 Seagen 达成 授权协议,获得了该药在除美国和加拿大之外全球其他国家的商业化权利。该 ADC 药物 由靶向 CD30 的嵌合 IgG1 单克隆抗体 cAC10 和一种微管蛋白抑制剂(单甲基 auristatin E, MMAE)通过一种蛋白酶敏感的可剪切连接子随机偶联而成,其中 CD30 是 HL 和 sALCL 的明确标志物。淋巴瘤患者预后差,对于复发患者治疗措施更为有限,存在较大的未满足 临床需求。
Adcetris 治疗复发或难治性 cHL 主要临床终点 ORR 为 75%。在一项全球多中心、 开放标签、II 期研究中(NCT00848926),102 例自体干细胞移植后复发或难治性 cHL 患 者接受每 3 周一次的 Adcetris 静脉给药(1.8mg/kg),主要临床终点为放射性诊断设备的 客观缓解率(ORR)。结果显示,在 94%患者中检测到肿瘤缩小,ORR 为 75%(95%CI, 64.9%-82.6%),CR 为 34%(95%CI,25.2%-44.4%),总体疾病控制率 DCR 高达 96% (90.3%-98.9%)。
Adcetris 治疗复发或难治性 cHL 次要临床终点 mPFS 为 5.6 个月,12 个月 OS 为 89%。在肿瘤有所缩小的患者中,mDOR 为 6.7 个月(95%CI,3.6-14.8 个月)。在完全 缓解(CR)的患者中 DoR 为 20.5 个月(95%CI,10.8 个月-NE)。截止数据分析日,102 个患者中 28 个死亡,总体 12 个月生存率为 89%(95%CI,83%-95%)。所有患者的评估 mPFS 为 5.6 个月(95%CI,5.0-9.0 个月),而对于 Adcetris 治疗后完全缓解患者(CR), mPFS 长达 21.7 个月,无疾病进展生存期较无 CR 的患者显著延长。
Adcetris 治疗复发或难治性 sALCL 主要临床终点 ORR 为 83%。一项评估 Adcetris 单药治疗复发或难治性 sALCL 患者的多中心、开放标签、II 期研究中(NCT00866047), 共 58 名患者入组,每三周用药一次(1.8mg/kg)。临床结果显示,总体 ORR 为 86%(95%CI, 74.6%-93.9%),完全缓解占 57%(95%CI,43.2%-69.8%),部分缓解占 29%。在 97% 患者中检测到肿瘤缩小。研究者评估的 ORR 为 83%(95%CI,70.6%-91.4%),CR 为 60%(95%CI,46.6%-73%)。mPFS 为 13.3 个月(95%CI,6.9 个月-NE)。截至数据分 析日,18 个患者死亡,mOS 尚未成熟,12 个月生存率为 70%。
ECHELON-1 和 ECHELON-2 两项 III 期临床试验结果奠定 Adcetris 针对 cHL 和 sALCL 的一线治疗地位。基于上述两项数据令人惊艳的临床试验,2011 年 8 月,Adcetris 获得 FDA 加速批准用于干细胞移植失败或至少 2 种多药化疗方案治疗失败且不适合干细 胞移植的 HL 患者,以及至少 1 种多药化疗方案治疗失败的 sALCL 患者,成为自 1977 年以来首个获批治疗 HL 和 sALCL 的新药。在随后的研发过程中,Adcetris 适应症不断扩大, 改变了HL和sALCL的临床治疗格局。2020年ASH大会公布ECHELON-1和ECHELON-2 两项 III 期临床试验的五年更新结果,与标准化疗方案相比,Adcetris 联合化疗方案对于一 线治疗晚期 HL 和 CD30 阳性外周 T 细胞淋巴瘤患者展现出更具优势的 PFS,奠定其作为 较一线标准化疗更优选择的临床地位。
截至2021年12月,Adcetris在全球超过75个国家获批上市,适应症多达6个。Adcetris 于 2020 年 5 月获得 NMPA 批准在中国上市,用于治疗复发或难治性 sALCL 或 CD30 阳 性 HL 成人患者。2020 年 Adcetris 全球销售额高达 6.59 亿美元,同比增长 5%,2021H1 全球销售为 3.45 亿美元,同比增长 4%。
Kadcyla:全球商业化最成功的 ADC 药物
Kadcyla 系全球首款用于治疗实体瘤,也是国内首个上市的 ADC 药物。Kadcyla (ado-trastuzumab emtansine, T-DM1)是罗氏制药公司继赫赛汀(曲妥珠单抗)和帕捷特 (帕妥珠单抗)之后推出的第三款 HER2 产品,进一步巩固其 HER2 霸主地位。2013 年, Kadcyla 获得 FDA 批准上市,成为首款治疗实体瘤的 ADC 药物,也是首个靶向 HER2 的 ADC 产品。Kadcyla 由靶向 HER2 的赫赛汀与 Immunogen 公司提供的微管蛋白抑制剂美 坦新(DM1)通过稳定的不可剪切连接子 MCC 连接而成。Kadcyla 有两项适应症在全球 获批:(1)在 100 多个国家/地区被批准作为单一药物,治疗先前接受赫赛汀和紫杉烷类 化疗(单独或联合)的 HER2 阳性晚期乳腺癌患者(2)Kadcyla 在美国和欧盟被批准用 于新辅助治疗后有残留病灶的 HER2 阳性早期乳腺癌患者的辅助(手术后)治疗,该适应 症 2020 年 1 月获得 NMPA 批准上市后,成为中国首个获批上市的 ADC 药物。
Kadcyla 对比赫赛汀用于早期乳腺癌患者辅助治疗取得优效结果。Kadcyla 国内上市 申请获批主要基于随机、开放标签、多中心 II 期 KATHERINE 研究的结果(主要临床终点 iDFS)。KATHERINE 共招募 1486 例新辅助疗法治疗后在乳腺和/或腋窝淋巴结中仍有病 理性侵袭性病灶残留的 HER2 阳性的早期乳腺癌患者,在术后以 1:1 随机分组,分别接受 Kadcyla 或赫赛汀作为辅助治疗。该研究旨在比较 Kadcyla 和赫赛汀作为术后辅助治疗对于这些新辅助治疗后有病灶残留的 HER2 阳性的早期乳腺癌患者的疗效和安全性。结果显 示,治疗 3 年后,Kadcyla 和赫赛汀两组患者主要终点无浸润性生存率(iDFS)分别为 88.3% 和 77.0%, Kadcyla 与赫赛汀相比,iDFS 显著更高,复发或死亡风险降低 50%。
Kadcyla 对比化疗延长患者无进展生存期 9.6 个月,NCCN 指南将其列为 HER2 阳性乳腺癌二线治疗首选方案。另外两项 III 期临床试验 EMILIA 和 TH3RESA 评估 Kadcyla 单 药治疗 HER2 阳性晚期乳腺癌患者中的有效性和安全性。EMILIA 是一项全球多中心、随 机、开放标签的 III 期临床试验,入组患者为 991 例既往赫赛汀和紫杉类治疗失败的 HER2 阳性局部晚期或转移性乳腺癌患者,按 1:1 随机分配,接受 Kadcyla 或对照药物(卡培他 滨和拉帕替尼)。试验结果表明,Kadcyla 组和对照组主要终点中位 PFS 分别为 9.6 和 6.4 个月(HR=0.65),次要终点中位 OS 分别为 30.9 和 25.1 个月(HR=0.68),ORR 分别为 43.6%和 30.8%。安全性分析与既往报告相似。基于此,NCCN 指南将 Kadcyla 列为不可 切除或转移性 HER2 阳性乳腺癌二线治疗首选方案。
Kadcyla 治疗三线及以上晚期乳腺癌患者较化疗方案 PFS 及 OS 显著获益。 TH3RESA 为 Kadcyla 与研究者选择的化疗头对头比较治疗 HER2 阳性转移性乳腺癌的多 中心、随机、开放标签 III 临床研究,共纳入 602 例既往接受过至少 2 种 HER2 靶向治疗 的晚期乳腺癌患者,随机分为 Kadcyla 组和研究者选择的化疗组(分组比例 2:1)。结果显 示,Kadcyla 的中位 PFS 优于研究者自选的化疗方案(6.2 个月 vs. 3.3 个月,HR=0.528), Kadcyla 组中位 OS 延长将近 7 个月(22.7 个月 vs.15.8 个月,HR=0.68)。同时 Kadcyla 组展示出更高的安全性,3 级以上的不良反应率为 40%(对照组为 47%)。该临床试验显 示 Kadcyla 组患者 PFS 和 OS 的显著获益,展示出 Kadcyla 在多线 HER2 阳性晚期乳腺 癌患者治疗的优异临床数据。
Kadcyla 联用免疫疗法的临床试验正在积极开展。Kadcyla 在单药治疗乳腺癌取得成 功之后也开展了与免疫疗法联用的临床试验。II期临床试验 KATE2比较 Kadcyla+Tecentriq 联用与 Kadcyla 单药治疗晚期 HER2 阳性乳腺癌的临床疗效,两组之间 PFS 无明显差异 且联用组副作用更为明显。近期,罗氏开展一项全球多中心、随机、双盲的 III 期临床试验 (KATE3 研究),旨在评估 Kadcyla+Tecentriq 联用与 Kadcyla 单药用于治疗 HER2、PD-L1 双阳性晚期乳腺癌患者的安全性和有效性。
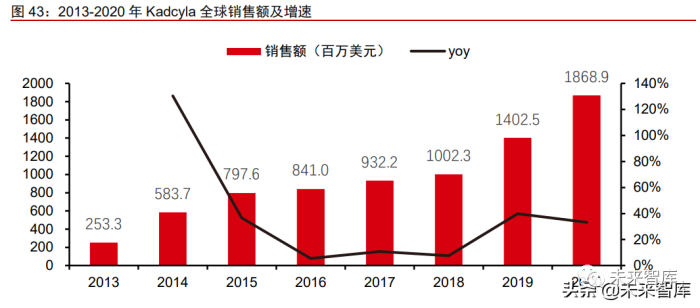
得益于优异的临床疗效,Kadcyla 成为目前全球商业化最成功的 ADC 药物。2020 年 全球销售额达 18.69 亿美元,同比增长 33%,在 2020 年度全球最畅销肿瘤药物中位列第 十一。
Enhertu:多癌种治疗展现非凡实力
Enhertu是已获批 ADC药物中研发管线布局最广的药物,被称为抗癌“神药”。Enhertu (trastuzumab deruxtecan,DS-8201)是利用第一三共开发的一款第二代 ADC 药物,由 靶向 HER2 的人源化 IgG1 单克隆抗体曲妥珠单抗(trastuzumab)、 公司创新合成的新型 拓扑异构酶 1 抑制剂 exatecan 衍生物(DX-8951 衍生物,DXd)和可剪切 4 肽链接子三 部分组成。2019 年 3 月,第一三共与阿斯利康达成一项总额高达 69 亿美元的免疫肿瘤学 合作,共同开展全球范围内 Enhertu 多个适应症的开发和商业化。Enhertu 是目前已上市 ADC 药物中研发管线布局最广的药物,在乳腺癌、胃癌、结肠癌、肺癌等多个领域都展现 出非凡的治疗潜力,被称为抗癌“神药”。目前,Enhertu 有两个 FDA 获批的适应症:HER2 阳性不可切除或转移性乳腺癌三线及以上;HER2 突变的胃或胃食管交界腺癌二线治疗, 成为该癌种获批的首个 ADC 药物。
阿斯利康财报公布, Enhertu(2019 年 12 月 FDA 获批上市)仅上市一年,在 2020 年的销售额就达 9600 万美元,2021 年仅三季度单季度销售额接近 6000 万美元,增长迅 猛。目前 Enhertus 针对多个癌种的临床正在快速推进,未来随着获批适应症的增多及上 市地区范围的扩大,Enhertu 的销售额还有巨大的上升空间。
乳腺癌
Enhertu 临床数据优异获 FDA 加速批准上市,弥补 HER2 阳性患者三线标准治疗空白。乳腺癌是 Enhertu 最先布局的癌种。DESTINY-Breast01 是一项开放标签、单臂、多 中心的 II 期临床研究,评估 Enhertu 在经 2 种或多种抗 HER2 靶向方案治疗后进展的 HER2 阳 性 转 移 性 乳 腺 癌 成 人 患 者 中 的 疗 效 和 安 全 性 。2019 年 12 月 21 日,基于 DESTINY-Breast01 研究,Enhertu 用于 HER2 阳性乳腺癌的后线治疗获得 FDA 加速批准 上市。2020 SABCS 大会上公布的最新结果显示,中位随访时间 20.5 个月后,ORR 达 61.4% (2019 SABCS 公布经 ICR 确认的 ORR 为 60.9%),相比较于其他 HER2 阳性晚期乳腺 癌二线治疗的临床研究中 ORR 约 40%-50%,Enhertu 的疗效非常显著。mPFS 长达 20.8 个月,初步 mOS 为 24.6 个月(OS 数据成熟度约 35%),12 个月时 OS 率为 85%,18 个月时 OS 率 74%。
安全性评估方面,最常见的 3 级及以上不良事件包括中性粒细胞减少(20.7%)、贫血 (8.7%)和恶心(7.6%)。
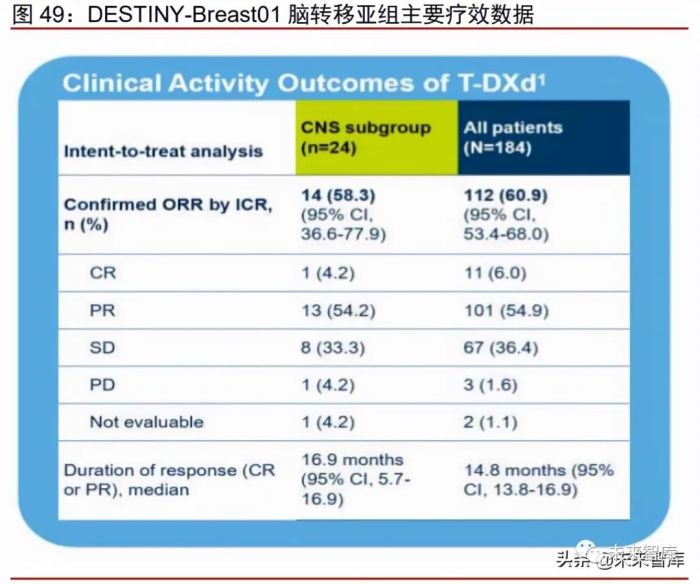
Enhertu 对 HER2+乳腺癌伴脑转移患者同样有效。对于 HER2+乳腺癌伴脑转移的患 者,目前的治疗措施仍然十分有限。2021 年 ASCO 大会公布研究者对 DESTINY-Breast01 队列中伴有脑转移的患者的亚组分析,结果显示经 ICR 确认的 ORR 高达 58.3%,中位缓 解持续时间为 16.9 个月,体现了 Enhertu 在 HER+乳腺癌伴脑转移患者中优异的疗效。目 前,公司已开展 4 项临床研究,用于 Enhertu 在 HER2+乳腺癌伴/不伴脑转移患者中的疗 效 评 估 :DESTINY-Breast07 (NCT04538742), DESTINY-Breast12 (NCT04739761), TUXEDO-1 (NCT04752059), DEBBRAH (NCT04420598)。
Enhertu 与 Kadcyla 针对二线 HER2 阳性乳腺癌头对头 III 期临床取得优效性结果。 目前,Kadcyla 是 NCCN 治疗指南中不可切除或转移性 HER2 阳性乳腺癌二线治疗首选方 案,而 Enhertu 获批上市的适应症为三线及以上治疗 HER2 阳性不可切除或转移性乳腺癌。2021年 ESMO大会,第一三共/阿斯利康公布了 Enhertu 与 Kadcyla头对头二线治疗HER2 阳性晚期乳腺癌的 III 期 DESTINY-Breast03 研究的中期分析结果。该研究共招募 524 例 患者,在分别随访 15.5 个月和 13.9 个月后,Enhertu 组尚未达到主要终点中位 PFS, Kadcyla 组中位 PFS 为 6.8 个月,Enhertu 将疾病进展或死亡风险降低 72%;亚组分析也 显示出一致的 PFS 获益,包括伴发脑转移患者;次要终点 OS 数据尚未成熟,但 Enhertu 组呈现出明显改善的趋势;与 Kadcyla 组相比,Enhertu 组 ORR 提高一倍多(79.7% vs 34.2%)。安全性方面,两组数据相当,Enhertu 安全性与既往数据一致。该结果有望使 Enhertu 替代 Kadcyla,成为赫赛汀和紫杉烷类经治后复发的 HER2 阳性转移性乳腺癌患 者的新治疗标准
Enhertu 在乳腺领域不断拓展,未来更多优异临床数据读出值得期待。 除 DESTINY-Breast01 外,Enhertu 在乳腺领域不断拓宽布局,探索 HER2 低表达和 HR+患 者中的疗效;用药方案从最初的单药治疗到如今开启联合方案的尝试。治疗线数逐渐前移, 从晚期后线开拓到辅助治疗。6 月 14 日,DESTINY-Breast09 完成首例患者给药。该项目 是一项全球范围、头对头的 III 期临床试验,评估 Enhertu 单药或者联合帕妥珠单抗与标准 治疗(紫杉烷、曲妥珠单抗和帕妥珠单抗)用于一线治疗 HER2 阳性乳腺癌患者的安全性 和有效性研究。这是 Enhertu 的首个评估一线治疗 HER2 阳性转移性乳腺癌患者的临床试 验,期待 Enhertu 在与标准疗法的比较中表现出更出色的治疗效果。
胃癌
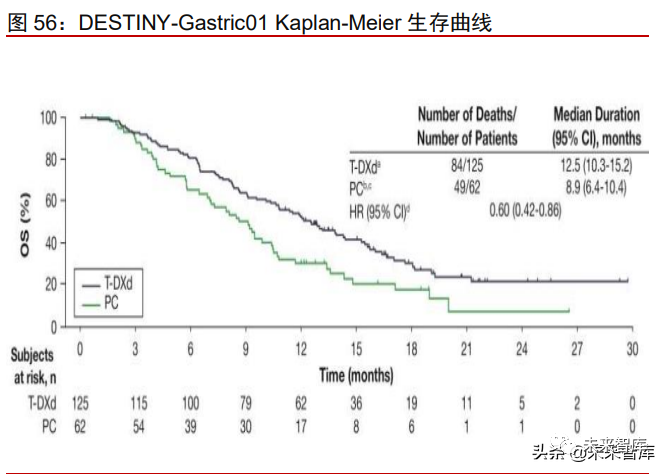
Enhertu 优于化疗方案,成为 HER2+胃癌首个获批的 ADC 药物。基于一项开放标签、 多中心、II 期临床试验 DESTINY-Gastric01 的研究结果,Enhertu 被 FDA 批准上市用于治 疗 HER2 突变的胃或胃食管交界腺癌,成为 HER2+胃癌首个获批的 ADC 药物。该研究对 比Enhertu和研究调查员选择的化疗用于治疗HER2阳性的不可切除/转移性胃或胃食管交 界处腺癌患者的有效性和安全性。截止至 2021 年 6 月,试验结果显示:在 175 例可评估 患者中,Enhertu 组 ORR 为 40.0%,远高于化疗组 12.5%。Enhertu 组的中位 OS 为 12.5 个月,化疗组为 8.9 个月,133 例达 OS 事件数,成熟度为 71.1%。Enhertu 组与化疗组 相比死亡风险降低 40%(HR=0.60;95%CI:0.42-0.86),充分说明 Enhertu 抗肿瘤活性优 于化疗方案。两组发生≥3 级不良事件的概率分别为 85.6%和 56.5%,Enhertu 组副作用 较对照组更高。
评估 Enhertu用于三线治疗HER2阳性晚期胃癌的 III期临床试验 DESTINY-Gastric01 已完成,目前公司正在开展多项临床试验,探索 Enhertu 在胃癌一线和二线治疗适应症中 的临床疗效。
结直肠癌
Enhertu 治疗 HER2 阳性转移性结直肠癌疗效显著。DESTINY-CRC01 是一项开放标 签、多中心、II 期临床试验,旨在评估 Enhertu 治疗既往接受≥两线标准治疗后出现进展 的 HER2 阳性、不可切除和/或转移性结直肠癌患者的有效性和安全性。受试者根据免疫组 织化学(IHC)和荧光原位杂交(FISH)测得的 HER2 表达量被分至三个队列:A. HER2 IHC3+或 IHC2+/ISH+;B. HER2 IHC2+/ISH2-;C.HER2 IHC1+。2021 ASCO 公布的数 据结果显示,A 队列共 53 例患者,中位随访时间为 62.4 周,经 ICR 确认的 ORR 为 45.3%, 疾病控制率(DCR)为 83.0%,中位缓解时间为 7.0 个月,中位 PFS 和中位 OS 分别为 6.9(4.1-8.7)和 15.5(8.8-20.8)个月。
目前无 HER2+结直肠癌治疗药物获批,Enhertu 为该患者人群提供潜在新选择。图 卡替尼(Tukysa)是一款抗 HER2 的小分子酪氨酸激酶抑制剂。2019 ESMO 公布一项图 卡替尼联合曲妥珠单抗治疗 HER2 阳性 RAS 野生型晚期直肠癌患者临床研究 ( MOUNTAINEER ) 的 初始结果 , 数 据 显 示 23 例 患 者 ORR 为 52.2%(95%CI: 30.6%-73.2%),中位 PFS 和中位 OS 分别为 8.1(95% CI: 3.8-NE)和 18.7(95% CI: 12.3-NE)个月。该结果是目前公布的 HER2 阳性晚期结直肠癌临床试验中疗效最显著的 一项,Enhertu 仅略逊于该方案。目前该适应症尚无获批的特异性药物,Enhertu 有望成 为治疗困境的新突破口。
安全性方面,Enhertu 在 DESTINY-CRC01 中出现的不良事件不容忽视。其中,任 意等级的间质性肺炎(ILD)发生概率为 9.3%(n=8)。ILD 是一种严重的不良反应,该 8 例患者确诊 ILD 后接受糖皮质激素治疗,除 4 例患者(Grade 2)病愈外,其余 4 例均出 现致死事件。研究者提出需要通过影像学检查等方法对受试者进行仔细监测,当出现可疑 ILD 时及时予以糖皮质激素干预。
肺癌
Enhertu 治疗 HER2 过表达 NSCLC 患者 ORR 为 24.5%。DESTINY-Lung01 是一项 开放标签、多中心的 II 期临床试验,旨在评估 Enhertu 用于治疗 HER2 过表达或突变的不 可切除可/或转移性非小细胞肺癌(NSCLC)患者的疗效。WCLC 2020 和 ESMO 2021 分别 更新了 HER2 过表达队列和 HER2 突变队列的研究结果。WCLC 2020 公布数据显示,截 止至 2020 年 5 月,HER2 过表达 NSCLC 患者队列共 49 位患者接受 Enhertu 治疗,ICR 确认的 ORR 为 24.5%(95% CI: 13.3%-38.9%),DCR 为 69.4%,中位 PFS 为 5.4 个月。其中 IHC2+和 IHC3+患者 ORR 分别为 25.5%和 20.0%。
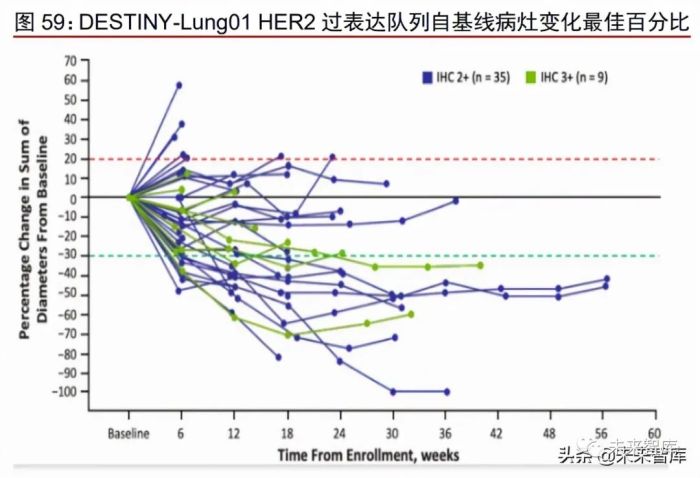
Enhertu 治疗 HER2 突变 NSCLC 获得 FDA 突破性疗法认定,更新数据显示 ORR 为 54.9%。ASCO 2020 公布了 Enhertu 治疗 HER2 突变 NSCLC 患者的疗效结果,42 位 患者中 ICR 确认的 ORR 达到 61.9%,DCR 为 90.5%,中位 PFS 为 14.0 个月,Enhertu 由此获得 FDA 授予的用于治疗 HER2 突变转移性 NSCLC 的突破性疗法认定。ESMO 2021 公布的更新数据显示,截至 2021 年 5 月,HER2 突变队列中,91 位患者接受 Enhertu 治疗,ICR 确认的 ORR 达到 54.9%(95% CI: 44.2%~65.4%),DCR 为 92.3%,中位 PFS 为 8.2 个月。
5 国内领军企业崛起,ADC 研发热潮正当时
荣昌生物:国内 ADC 赛道领头羊
荣昌生物上市国内首款自主研发 ADC 产品,ADC 药物领域国内领先。荣昌生物成立 于 2008 年,是一家具有全球视野的创新型生物制药企业,专注于 ADC、抗体融合蛋白、 双抗、单抗等抗体药物的研发和商业化。截止至 2021 年 6 月,公司已开发了近 20 款候选 生物药产品,其中 10 余款候选生物药产品处于商业化、临床研究或临床研究申请(IND) 准备阶段,均为靶向生物创新药。公司有两款核心产品获批上市:(1)爱地希(维迪西妥 单抗,RC48)是中国首款自主研发的 ADC 药物,也是目前唯一一款获得 FDA 突破性疗 法认定的国产 ADC 产品;(2)泰它西普(RC18)是全球同类首创的重组 B 淋巴细胞刺激 因子/增殖诱导配体双靶点融合蛋白型生物制剂,用于治疗系统性红斑狼疮。爱地希和泰它 西普双双进入 2021 年国家医保药品目录,将显著提高产品可及性,有利于获得更多的市 场份额和经济效益。
公司是国内 ADC 药物布局较广,研发进度最快的公司。爱地希于 2021 年 6 月 9 日 在中国获批上市,用于至少接受过 2 种系统化疗的 HER2 过表达局部晚期或转移性胃癌患 者的治疗,另一个适应症 HER2 表达晚期尿路上皮癌获得 FDA 和 CDE 突破性疗法认定, NDA 已于近期获得 NMPA 批准。此外,公司就爱地希开展 HER2 阳性适应症差异化布局, 包括胆道癌、非小细胞肺癌、乳腺癌。2021 年 6 月 29 日,CDE 公布爱地希纳入突破性 治疗品种,适应症为既往接受过曲妥珠单抗和紫杉类药物的 HER2 阳性存在肝转移的晚期 乳腺癌患者,这是爱地希第三次获得突破性疗法认定,充分展现该药巨大的治疗潜力。
公司 ADC 药物研发管线丰富,储备雄厚。除爱地希之外,公司还有 3 款临床阶段 ADC 药物,处于临床 I 期或 IND 阶段,包括 RC88、RC108、RC118,分别靶向于 MSLN (CAK1)、 c-MET 和 Claudin 18.2。其中,RC88 是靶向于 MSLN 的 ADC 药物中进展最快的一款, 有望成为国内同类首创产品。RC118Ⅰ期临床试验于 7 月 30 日获得澳洲伦理委员会许可, 这是荣昌生物首次获批在澳洲开展临床试验。
爱地希(维迪西妥单抗,disitamab vedotin, RC48)
爱地希相较同类竞品的竞争优势显著。爱地希是由新型人源化 IgG1 HER2 抗体、小 分子毒素 MMAE、可剪切连接子三部分组成。相较于同类竞争产品,爱地希有独特的分子 结构优势,主要包括拥有自主知识产权的比标准治疗手段(曲妥珠单抗)相比对 HER2 具 有更高亲和力的新型人源化抗体、高效的细胞内吞效应以及无溶酶体耐药性可裂解的连接 子、以及较强的“旁观者效应”。
爱地希治疗三线及以上胃癌患者,主要临床终点 ORR 为 24.4%。爱地希获批上市是 基于一项开放、多中心、单臂 II 期临床研究,共招募了 127 名曾接受过至少两次化疗治 疗的 HER2 过表达胃癌或胃食管结合部腺癌患者,每两周接受一次 2.5mg/kg 的爱地希治 疗。截至 2020 年 6 月 22 日的阶段性临床数据分析,经 ICR 确认的 ORR 为 24.4% (95%CI:17.2%-32.8%), 中位 PFS 为 4.1 个月(95%CI:3.5-4.8),中位 OS 为 7.6 个月(95% CI: 6.6-9.0)。安全性方面,治疗相关的不良事件主要包括白细胞计数降低(53.5%),脱发 (52.8%),中性粒细胞计数降低(49.6%)及乏力(45.7%),显示出较好的风险/收益比。作为维迪西妥单抗用于治疗胃癌在中国完全获批的条件,公司于 2020 年 9 月启动了爱地 希用于治疗胃癌的 III 期确证性临床试验。
爱地希治疗二线及以上尿路上皮癌,第一项 II 期临床试验 ORR 为 51.2%。公司开展 了一项爱地希用于治疗 HER2 过表达(IHC 2+或 IHC 3+)转移性或不可切除尿路上皮癌 的开放标签、多中心、单臂 II 期临床试验,共招募 43 人(其中 32.6%曾接受过至少两种 化疗,20.9%曾接受过免疫治疗)。该项目于 2021 年 5 月完成数据分析,结果显示,ORR 为 51.2%(n=22),DCR 为 90.7%(n=39),其中部分缓解患者有 22 名。中位 PFS 为 6.9 个月,中位 OS 为 13.9 个月。亚组分析表明,对于不同类型的转移性尿路上皮癌和接受过 PD-1/PD-L1 治疗患者,爱地希具有持续强效的抗肿瘤作用。安全性方面,最常见的 TRAE 为感觉减退(60.5%)、脱发(55.8%)、乏力(44.2%),显示出良好的患者耐受性。
爱地希治疗二线及以上尿路上皮癌,第二项 II 期临床试验 ORR 为 50%。根据该试验 结果,公司启动了一项单臂、多中心、注册性 II 期临床试验,以评估爱地希单药治疗在既 往一线化疗失败后的 HER2 过表达局部进展或转移性尿路上皮癌的有效性和安全性。本研 究入组 64 名患者(85.9%曾接受过治疗两种化疗,29.7%曾接受过免疫检查点抑制剂治疗), 截至 2021 年 3 月,试验结果显示独立影像学评估 ORR 达到 50%,DCR 为 76.6%,中位 PFS 为 5.1 个月,中位 OS 为 14.2 个月。该结果于 2021 年 ASCO 大会发布,充分证实 爱地希对于二线及以上治疗 HER2 阳性晚期尿路上皮癌患者有突出的疗效和生存获益。
爱地希联合特瑞普利单抗治疗尿路上皮癌患者(无需 HER2、PD-1 表达情况筛选)一线治疗 ORR 高达 100%。另一项爱地希联合 PD-1 抗体拓益(特瑞普利单抗)治疗局部 晚期或转移性尿路上皮癌的单臂 Ib/II 期研究初步结果也在 2021 年 ASCO 大会上首次公布。17 例患者完成了至少一次的疗效评价,初步结果显示整体 ORR 为 94.1%(16/17),其中 一线治疗患者 ORR 达 100%(10/10)。88.2%的患者在 8±1 周疗效评价时出现缓解。该 结果证实了 ADC 药物联合免疫疗法治疗理念的科学性,在无需进行 HER2 表达、PD-1 表 达等生物标记物筛选情况下,实现尿路上皮癌治疗领域的突破,有望成为一线尿路上皮癌 治疗的新选择。
爱地希针对 HER2+肝转移晚期乳腺癌患者获突破性疗法认定,mPFS长达 1 年以上。 在扎堆严重的适应症 HER2 阳性乳腺癌领域中,爱地希也突出重围,差异性布局用于治疗 既往接受过曲妥珠单抗和紫杉类药物的 HER2 阳性存在肝转移的晚期患者,对于这类患者 治疗指南尚无推荐的药物选择。此次突破性疗法认定是基于一项随机、对照、多中心 II 期 临床试验,旨在评估爱地希单药治疗对比帕替尼联合卡培他滨的疗效和安全性,适应症为 既往接受过曲妥珠单抗和紫杉类药物的 HER2 阳性乳腺癌伴肝转移患者。结果显示,爱地 希对比拉帕替尼联合卡培他滨则显示出了较高的有效率(63.2% vs 39.5%),同时也显示出良好的生存获益,中位无进展生存期达 1 年以上(12.5 个月 vs 5.6 个月)。
HER2 阳性和低表达乳腺癌患者均能从爱地希治疗中获益,公司已启动 HER2 低表达乳腺癌患者的 III 期临床。2021 年 ASCO 大会上,公司披露爱地希治疗 HER2 阳性和 HER2 低表达的晚期或转移性乳腺癌患者的 I 期和 Ib 期两项临床试验的合并分析结果。两项临床 试验共招募 70 名 HER2 阳性乳腺癌患者,主要试验终点为评估爱地希的最大耐受剂量、 安全性和明确 II 期临床试验的推荐剂量;Ib 期临床试验 48 名 HER2 低表达乳腺癌患者, 主要试验终点为探索有效剂量和确定 II 期临床试验。基线时,47 例患者(39.8%)既往 已接受过至少 3 线化疗。结果显示,HER2 阳性和 HER2 低表达的乳腺癌患者都能够从爱 地希治疗中获得较好的疗效,且未出现新的安全性问题,2.0mg/kg 剂量组的获益风险比 最佳。基于Ib期的结果,公司已和CDE沟通启动一项治疗HER2低表达(IHC2+ 且FISH-) 乳腺癌患者的 III 期注册性临床试验。
爱地希授权 Seagen 获最高 26 亿美元收入总额,彰显荣昌生物在全球 ADC 领域的重要地位。2021 年 8 月 8 日,荣昌生物与国际知名生物制药企业 Seagen 公司达成一项全 球独家许可协议,Seagen 公司将获得爱地希在亚洲区域(不包括日本、新加坡)以外的 全球开发和商业化权益。荣昌生物从此次交易中获得的潜在收入总额将高达 26 亿美元, 包括 2 亿美元首付款和最高可达 24 亿美元的里程碑付款,刷新中国单药海外授权交易的 最高纪录。该合作协议充分显示出爱地希的巨大商业价值以及荣昌生物在全球 ADC 领域 的重要地位。
荣昌生物获资本市场青睐,充沛现金将支撑公司在 ADC 领域的不断开发。荣昌生物于 2019 年开始第一轮融资,在上市前已完成 6 次融资,为企业研发能力及资金提供良好 背书。2020 年 11 月 9 日,荣昌生物成功登陆港交所主板,吸引了由高瓴资本等 19 家投 资机构组成的超豪华基石阵容,成为港交所 IPO 企业中基石投资者最多的新股之一。募资 5.9 亿美元,创造了 2020 年全球生物医药 IPO 募资最高纪录。近期,荣昌生物拟募资 40 亿元赴科创板上市,所得资金中接近一半将用于泰它西普、爱地希和 RC28 三款重磅产品 的开发和商业化。充沛的现金将有力支持荣昌生物在 ADC 领域的进一步发展。
乐普生物:致力于开发同类首创 ADC 药物
收购 ADC 药物研发能力强劲的上海美雅珂生物,跃升国内 ADC 赛道领军企业。乐普 生物成立于 2018 年,聚焦于 PD-1/PD-L1、ADC 药物和溶瘤病毒类等肿瘤治疗领域药物 和联合疗法的研发。通过投资并购、自主研发和合作引进,公司发展迅猛,现已搭建靶点 发现、成药研制、开发和生产的综合性产业平台,拥有厚德奥科、翰中生物、上海美雅珂、 上海航嘉孵化器等 9 个子公司。目前,公司产品管线中有 8 种临床阶段候选产品。公司于 2018 年收购翰中生物所得的 PD-1 产品普特利单抗上市申请近期获得 NMPA 受理,同年 收购 ADC 药物自主研发能力强劲的上海美雅珂生物,成为 ADC 赛道领军企业。2021 年, 乐普生物在完成 3 轮融资后向港交所递交上市申请。
布局 5 款 ADC 药物均处于临床阶段,其中 CMG901 为全球首个获批临床的靶向 CLDN18.2 的 ADC 药物。目前,乐普生物研发管线中共有 5 款 ADC 药物,包括 MRG003、 MRG002、MRG001、CMG901 和 MRG004A。随着 2021 年 8 月 MRG004A 临床申请获 得 NMPA 默示许可,公司的 5 款 ADC 药物均已进入临床开发阶段,形成独有的差异化 ADC 产品体系。MRG003、MRG001、CMG901 和 MRG004 分别靶向于 EGFR、CD20、 CLDN18.2,根据临床研究进展,这四款 ADC 候选药物有望成为国内同类首创 ADC 产品。其中,CMG901 由公司和康诺亚生物合作开发,是全球获得 IND 批准的首款 CLDN18.2 靶向 ADC 药物。
MRG003 有望克服 EGFR-TKI 耐药突变的临床难题。MRG003 由人源性 EGFR 单克 隆抗体、可剪切 VC 连接子和小分子毒素 MMAE 连接而成。EGFR-TKI(EGFR 酪氨酸激 酶抑制)已广泛应用于 EGFR 突变或异常表达的肿瘤患者,然而部分患者接受小分子 EGFR-TKI药物治疗出现获得性耐药突变,成为亟待解决的一大临床难题。MRG003强大、 广谱的抗肿瘤疗效在多种肿瘤小鼠模型(包括 CDX 模型和更具临床相关性的 PDX 模型) 研究中得到证实。例如,在对第三代 EGFR-TKI 奥希替尼具有耐药性的 NSCLC PDX 模型 LUN2210-4a 中测试 MRG003 的抗肿瘤活性,MRG003 呈现出剂量依赖性的抗肿瘤活性, 3mg/kg 剂量下在第 28 天彻底根除肿瘤。因此,MRG003 最大的临床优势是有望克服 EGFR-TKI 引起的多种获得性耐药突变,满足这类患者的临床需求。
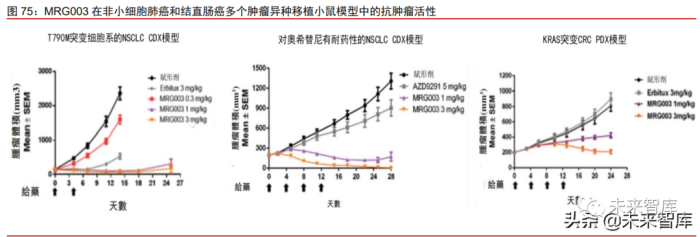
MRG003 多瘤种 I 期临床数据良好,四个适应症已进入国内 II 期临床。在一项晚期实 体瘤的开放标签、多中心 I 期临床研究中,Ia 期剂量爬坡阶段入组 22 例晚期实体瘤患者, Ib 期剂量扩展阶段入组 39 例 EGFR 阳性实体瘤患者,包括复发性头颈鳞状细胞癌 (HNSCC)、鼻咽癌(NPC)和结直肠癌(CRC)。临床数据表明 EGFR 阳性状态患者接 受有效剂量大于 1.5 mg/kg 的 MRG003 实现肿瘤缓解或肿瘤控制。Ib 期 27 例可评估患者 中(剂量 2.5mg/kg),整体 ORR 和 DCR 分别为 30.0%和 63.0%,其中 HNSCC(ORR 为 40%,DCR 为 80%)和 NPC(ORR 为 44%,DCR 为 78%)患者获益显著。安全性 方面,入组的 61 名患者中,14 名患者(23.0%,Ia 期 4 例及 Ib 期的 10 例)报告了 3 级 或以上的治疗相关不良事件。目前,MRG003 在国内有 4 条管线处于临床 II 期阶段,布局 HNSCC、NPC、NSCLC 和胆道腺癌(BTC)4 个适应症。
MRG001 治疗 CD20 阳性、美罗华耐药的 NHL 临床前体内研究数据亮眼。MRG001 是临床进度领先的 CD20 靶向 ADC 药物,由美罗华(利妥昔单抗)生物类似药、可裂解 肽键 vc-PABC 连接子和微管蛋白抑制剂 MMAE 组合而成,用于治疗 CD20 阳性复发或难 治性 B 细胞非霍奇金淋巴瘤(NHL)。目前,CD20 单克隆抗体美罗华已作为一线药物, 用于治疗难治性、复发性、初治 CD20 阳性 B 细胞 NHL。然而,约 30.0%-60.0%的初治 性患者在基线时对美罗华耐药;接受美罗华治疗后复发后,约 60.0%的患者对美罗华第二 疗程反应不佳。MRG001 旨在克服美罗华耐药问题,满足这类患者迫切的临床需求。
在临床前体内研究中,利用美罗华耐药的弥漫大 B 细胞性淋巴瘤(DLBCL)PDX 模 型,证实了 MRG001 出色的抗肿瘤活性,3mg/kg 剂量下在第 28 天彻底根除肿瘤。在该 特定模型中,与美罗华相比,MRG001 的抗肿瘤生长能力提高至少十倍。正在进行的开放 标签、多中心 I 期临床研究初步结果已显示出令人惊喜的疗效和良好的安全性。一名滤泡 性淋巴瘤患者在 0.15 mg/kg 的起始剂量下达到 PR,一名 DLBCL 患者在 1.8 mg/kg 的剂 量下达到 CR。发生的两起剂量限制毒性事件均在无医疗干预的情况下得以解决。
I 期研究结果显示 MRG001 在 NHL 患者中具有可管理的安全性和初步抗肿瘤活性。 目前,MRG001 正在进行一项开放性、多中心 l 期研究,评估其在 CD20 阳性复发或难治 性 B 细胞非霍奇金淋巴瘤(NHL)患者中的安全性、耐受性、药代动力学和初步抗肿瘤活 性。截止到 2021 年 5 月 31 日,共入组 21 例患者,包括 DLBCL(n=8)、滤泡淋巴瘤(FL) (n=12)和边缘区淋巴瘤(MZL)(n=1)。ASH 2021 公布结果显示,临床 II 期推荐剂量 (1.8mg/kg)有 6 例受试者(4 例 DLBCL 和 2 例 FL),其中 1 例 DLBCL 受试者达到 CR, 另一例 DLBCL 受试者达到 PR, ORR 为 33.33%(2/6),3 例达到 SD,DCR 为 83.3%。此研究表明,MRG001 在 NHL 患者中具有可管理的安全性和初步抗肿瘤活性。全球尚无 已上市的 CD20 ADC 产品,MRG001 作为复发/难治晚期 NHL 新药,临床进度领先,具有 FIC 的潜力。
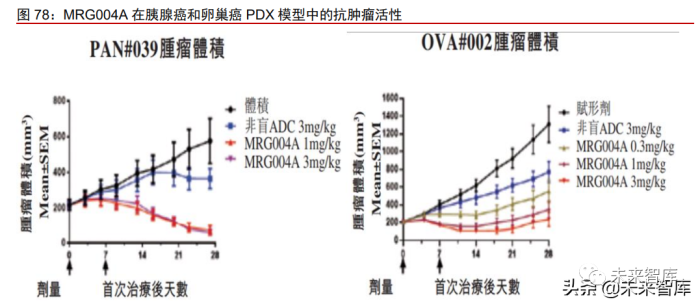
MRG004A 是一款靶向组织因子(tissue factor, TF)的 ADC 药物,全球在研项目稀少。TF 的异常表达与多种肿瘤进展、肿瘤转移和治疗预后关系密切,被认为是一个有吸 引力的癌症治疗靶点。全球范围内针对这一靶点的在研新药项目稀少,ADC 药物仅有 3 款,其中进展最快的 TF 靶向 ADC 药物是 Genmab 与 Seagen 联合开发的 tisotumab vedotin,于 2021 年 2 月向 FDA 提交上市申请。与 tisotumab vedotin 所利用的传统偶联 技术相比,MRG004A 运用从 Synaffix 公司引进的 GlycoConnect 定向偶联技术和 HydraSpace 极性间隔技术,其优势在于降低药物在血液循环中脱落风险和毒性,使药物 具有更宽的治疗窗口。
动物模型展示出强大抗肿瘤活性,系国内首个获批临床的 TF 靶向 ADC 药物。在临 床前研究中,MRG004A 在三阴性乳腺癌动物模型中比传统偶联 ADC 在血液循环中有更 好的稳定性和更强的药效,在胰腺癌及卵巢癌模型中也表现出强大的抗肿瘤活性。2021 年,该产品 IND 申请在国内和美国均获批临床,将开展针对 TF 阳性晚期或转移性实体瘤 的 I/II 期研究。MRG004A 是国内首个 IND 获批的 TF 靶向 ADC 药物,也是公司第 4 款有 望成为同类首创的候选产品。
多禧生物:技术平台优势显著
杭州多禧生物是一家成立于 2012 年的国家级高新技术企业,专注于 ADC 的研发、生 产、临床和商业化。经过近 8 年的沉淀,多禧生物建立了完善的 ADC 创制平台,在抗体、 连接子、小分子毒素三大元件中均展现出强大的核心能力。连接技术方面,拥有定点偶联 等新型智能化连接子约 50 余种;小分子细胞毒性化合物方面,拥有 5 大类约 100 余种分 子;抗体工程方面,建立了完善的抗体构建和生产工艺,确定了 24 个可用于后期抗体开 发的靶抗原。此外,公司还建立了完整的 ADC 药品开发与评价平台。公司拥有专利申请 超 300 项,仅在美国获得授权的专利就达到 20 项。凭借先进的技术平台和经验丰富的团 队,公司将在未来几年迎来收获期。
多禧生物是国内 ADC 药物布局最多的企业之一,拥有不同阶段的 17 款产品管线,其中有 4 款药物已进入临床试验阶段。
DAC-001(DX126-262)是公司自主研发的新一代ADC药物,由重组人源化HER2 单克隆抗体 DX-CHO9 通过半胱氨酸的巯基连接 Tubulysin B 类似物形成,其中 Tubulysin B 类似物是公司具有自主知识产权的高活性细胞毒素分子。DAC-001 已开展针对 HER2 阳性晚期/转移性乳腺癌、胃癌的 I 期研究。
DAC-002 / JS108 是一款 TROP2 靶向 ADC 药物,由 TROP2 单抗通过智能连接 子与微管蛋白 Tubulysin B 类似物偶联而成。2019 年,君实生物通过独占许可授 权方式从多禧生物获得许可使用 DAC-002,在除日韩以外的全部亚洲国家及地区 开展 DAC-002 的临床开发和商业化。多禧生物将获得 3000 万元人民币的首付款 和 226.8 万元人民币的样品制备费用,以及累计不超过 2.7 亿元人民币的里程碑 款。2020 年 9 月,DAC-002 / JS108 启动 I 期临床试验,招募 153 例晚期实体 瘤患者。目前,国内有关 5 款 TROP2 靶向 ADC 药物处于临床开发阶段,进展 最快的是云顶新耀以 8.35 亿美元授权引进的 Trodelvy,已向 NMPA 递交上市申 请。
多禧生物的第三款 ADC 药物 DAC-003(重组人源化抗 MUC1 单抗-Tub201 偶联 剂)IND 在 2021 年 6 月获得批准,成为国内首个进入临床阶段的 MUC1 ADC 产品。Mucin1(MUC1)是一种高度糖基化的跨膜蛋白,广泛分布并异常丰富地 表达于癌细胞表面,在肿瘤发生和转移等方面起到重要作用。MUC1 作为靶点的 药物开发仍处于早期,目前临床阶段仅有一款 MUC1 靶向 ADC 药物 M1231 处 于 I 期临床招募中。此外,齐鲁制药从韩国 Peptron 公司独家授权引进 MUC1 ADC 药物 Pab001-ADC,尚处于临床前阶段。多禧生物这款 DAC-003 差异化布局国 际前沿靶点,提升了企业的核心竞争力。
2021 年 12 月,多禧生物的第四款 ADC 药物 DXC007 获批临床,适应症为复发 或难治性 AML,靶点尚未公布。

本文来源于中信证券,,陈竹,刘泽序
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多