干货|药品注册现场核查法规及核查要点(附流程图)
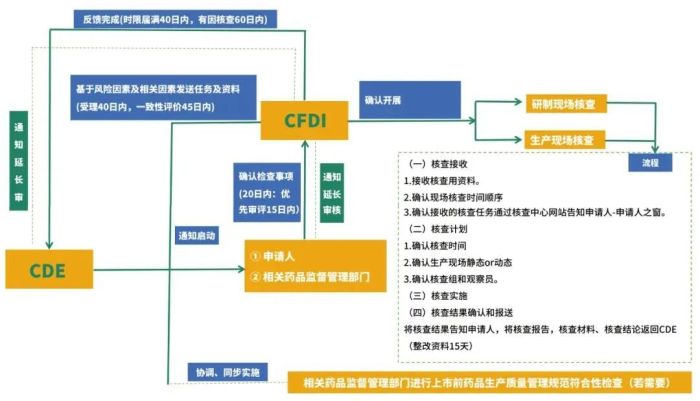
注册现场核查流程图
(点击查看大图)
备注:
核查过程中认为有必要进行样品检验的,所抽取的样品应在核查结束之日起10日内,送达指定药品检验机构。 延长时限不超过原时限的二分之一。
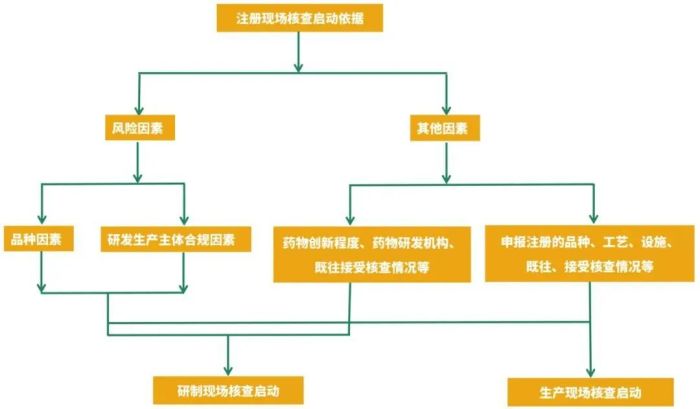
核查启动因素示意图
(点击查看大图)
备注:
原则上以品种因素和研发生产主体合规因素中风险情形较高的确定注册申请风险等级,特殊情况的除外。对于补充申请,生物制品和中药注射剂不纳入品种因素高风险情形。
研制现场核查内容 准备内容
药品研制基本情况(如属委托,应说明被委托研究单位基本情况)。
研制所涉及的批次(含BE批)批量用途,研制批次(含BE批)生产的地址、生产线、时间地点、使用量和剩余量等。
研制批次(含BE批)所用处方生产工艺、原辅料包装材料来源及标准、生产线(设备设施)、产品质量标准(含中间控制标准)等是否与已上市/拟上市商业化生产规模的批次一致。
参比制剂的来源、采购和使用情况。
药品和参比制剂体外研究的对比研究情况,研究时间、批号和研究结果。(质量对比和溶出曲线)
药品关键质量属性(含稳定性)研究情况。(全检数据及稳定性数据)
接受现场检查品种全套注册申报资料。
委托研究协议和质量协议,如有。
参比制剂的来源及证明,如购买发票、赠送证明等。参比制剂的包装标签、说明书、剩余样品等。参比制剂的接收、发放、使用记录或凭证。
药品相关研究记录,包括:处方工艺研究原始记录,如有;样品试制相关原始记录;质量研究相关原始记录;体外评价及稳定性研究的相关原始记录;仪器设备使用记录;纸质图谱及电子图谱。
其他相关文件,包括:处药品检验方法确认或验证资料;稳定性试验方案及报告;体外研究总结报告;溶出度仪的验证资料;研究用的剩余样品情况(不应销毁)。
核查要点
组织机构与人员:研究内容相适应的管理机构,相应的质量管理;具有资质的人员
研究条件:场地、设备、仪器和管理制度
文件和记录:应当建立文件和记录管理的制度或标准操作规程。药物研究开发全过程应有相应记录,包括预试验和探索性研究的数据和记录 变更和偏差管理:至少在药物进入临床阶段后就应当建立与药物研发阶段相适应的变更、偏差和失败管理制度或标准操作规程,针对关键批次出现的偏差或失败应当得到适当的调查和/或分析,并进行记录
委托机构: ➢ 委托其他机构进行全部或部分药学研究及样品试制的,委托方应当对受托方的研究能力、质量管理体系等进行评估,以确证其研究条件和研究情况 ➢ 双方应当签订委托合同或其他有效证明 ➢ 委托方应当对委托研究的过程和结果负责,并确保委托研究过程中的数据可靠性。受托方应当遵守相关要求,保证研究及样品制备过程规范、数据真实可靠、研制过程可追溯 判定标准(不合格情况):
发现真实性问题或申报资料真实性存疑,申请人不能证明其真实性的; 关键研究活动、数据缺少原始记录导致无法溯源的; 发现与申报资料不一致,可能影响质量评价的; 存在严重的数据可靠性问题,导致对药品安全性、有效性、质量可控性的评价产生影响的; 拒绝、不配合核查,导致无法继续进行现场核查的。


内容来源:
责任编辑:胡静 审核人:何发
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

AI+制药行业潜力巨大,产业链相关公司梳理(名单)
2026-04-29
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

解读2023版药品GMP指南中的检重仪精度要求
2026-05-08
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多