创新药药学研究的关注点
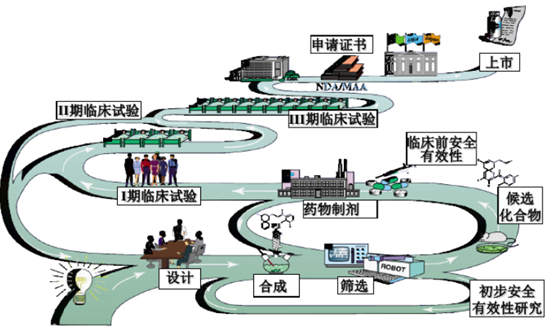
一个创新药的研究开发需要经历:疾病市场调研、分子机理研究、靶标的选择确认、分子设计、先导化合物合成及优化、药效药代和毒性初筛、临床前研究、临床研究、新药审评及上市等环节,从新药立项到最终上市,需要经历大约8-10年时间周期,所谓:“十年磨一剑”。新药药学研究经历了一个从发现到认知再到确认的过程,在这个过程中,安全性和有效性始终是筛选或淘汰候选药物的”金标准”,药学研究任务的顺利开展是支持后期临床前研究和临床研究的重要前提,是保证拟上市药物质量可控性的关键所在。
新药药学研究是一个充满不确定性及循序渐进的过程,其研究的深度和广度会随着新药开发的进程逐步推进,会逐步建立安全有效性与产品质量特性之间的关系。且在这个过程中,由于创新药不同研究阶段的药学研究目标不同,决定了变更亦是创新药研发过程中一个永恒的话题,变更可能会对临床进度、受试者的安全有效性或临床结果产生影响,引入一定的质量风险,所以需要药学研究人员充分评估变更可能引入的质量风险并全面开展相关研究,以支持这些变更应用于临床试验样品的制备。
处方前研究是药学研究工作的首要任务,其主要通过借鉴查阅国内外的文献资料或进行相应的实验研究,为后期剂型的选择、制剂的处方工艺研究提供依据。在制剂研究阶段,首先应对候选药物的物理性质、化学性质、生物学性质等一系列基本性质进行研究,其具体研究内容主要涉及有:药物的溶解度、渗透性、溶出速度、粒径大小、晶型、pKa电离常数、分配系数、引湿性、原辅料相容性、药物稳定性、药物的药动药效学等研究。基于QbD“质量源于设计”理念,处方前研究的另一重要目的是为制剂研究提供初步风险评估的依据,为制剂研究的方向和范围提供一个导向作用。
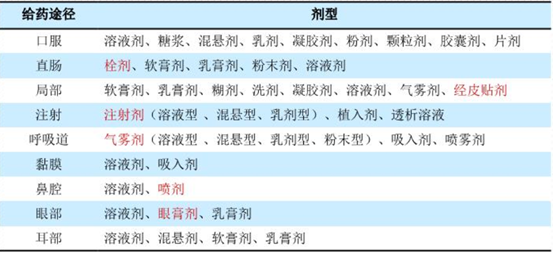
一般会根据药物的理化性质和治疗需求,确定给药的最佳途径,选择合适的剂型和规格。剂型设计的原则是基于安全、有效、质量可控、方便。所以药学研究人员会根据API的理化性质找出制剂研发的关键点,选择合适的剂型、辅料、制剂技术或工艺等,特别是对于一些BCSⅡ或Ⅳ类难溶解性药物,重点还是要根据药物的溶解度和稳定性来考虑适当的剂型开发。根据市场调研,口服给药占比在70%以上,因为口服给药溶出度较高、口服吸收好、适用于长期给药,使用方便、安全,故在剂型开发设计中是尽可能首选;注射给药适用于不能口服给药的病人,或药物在胃肠道不稳定、口服吸收效果不好,用于急救、重症或速效(如麻醉等);皮肤/粘膜给药可作用于身体不同局部部位,作用持久而稳定,如吸入或舌下给药还可发挥速效作用,使用更为安全有效、方便。
其次是规格,规格是指一个剂量单位中含活性成分的量。对于创新药而言,其规格的设计应该满足用量剂量和药剂学技术两个方面的基本要求。一般会结合早期与阳性药的药效比较数据以及安全性结果、考虑Ⅰ期爬坡剂量,由临床前、临床、药学三方共同讨论初步确定规格。在临床试验阶段,通常设计多个规格来满足对有效性剂量的探索,最终根据临床试验结果确定最佳的单次用药剂量或剂量范围,作为拟上市规格进行申报。
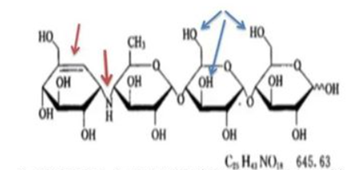
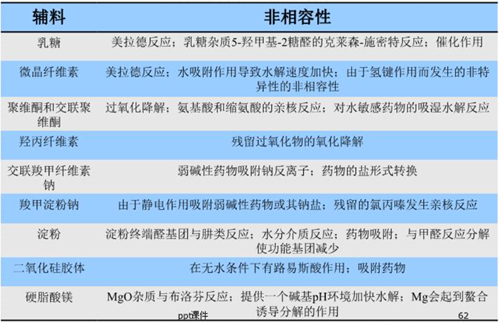
在制剂处方工艺研究方面,重点还是要关注制剂处方的稳定性、处方工艺的可放大性。对于制剂处方的稳定性考虑,在处方设计前最好能将可能存在的稳定性风险全面考虑在内,比如在原辅料相容性问题上,从API的结构出发,全面考虑可能与辅料产生的潜在杂质,例如阿卡波糖片结构中含有一个伯氨基,弱碱性,高水分下可以发生美拉德反应,最好不与乳糖配伍;双键不稳定,易发生加成反应、自由基反应,避免高水分、金属离子等。将风险在源头“扼杀”,才能确保研发工作的顺利推进。基于对处方工艺的可放大性的考虑,在制剂工艺的小试研究阶段,就尽可能选择与批量放大生产、商业生产相匹配的设备,比如在普通湿法制粒工艺小试阶段,使用流化床进行干燥以适应生产线,尽可能使用旋转压片机进行小试确认,这样可以有效降低临床备样的风险,而对于液体注射剂,注射剂配方尽可能简单,按照欧盟灭菌决策树开展研究。
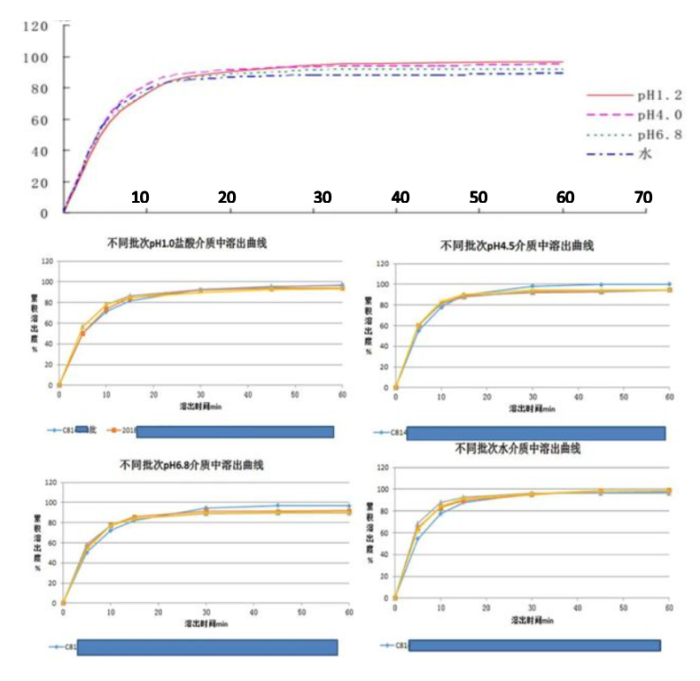
药学研发人员将API辅以一些辅料、采用适当的工艺制成固体制剂,如片剂或胶囊剂等,那接下来如何科学、有效的进行制剂处方/工艺/辅料的筛选?主要评价指标就是溶出度实验,由于药物本身的水溶性大小或溶解速率不同,或者制剂因素对药物溶解速率的影响等,使得溶出度有所差异。如何筛选溶出差异,就需要选择具有区分力的溶出介质,具有区分力的溶出介质可以筛选出关键质量属性、关键工艺参数下溶出方法的区分力,例如不同配方、不同工艺(硬度、崩解剂用量等)、不同API粒度等。临床前研究过程中,根据API的BCS分类及渗透性绘制溶解度-PH曲线至关重要,由该数据可以初步判定该制剂是否为PH依赖性制剂,从而指导创新药研发与溶出度质量标准的拟定。且在药学研究过程中,要积累多种PH介质的溶出曲线,积累关键批次的溶出曲线至关重要,通过溶出曲线的测定,可以评价生产工艺的稳定性;可以评价批间/批内样品的质量是否相一致;可以评价各类变更(如处方变更、工艺变更、生产规模变更、原辅料来源变更、生产场地变更等)是否会影响该制剂的内在质量。
例如某普通片剂的PH-溶解度依赖性曲线得知其无PH依赖性,不同批次样品在四种介质中无明显差异,批间/批内样品内在质量相一致。
在创新药药学研究过程中,要实时关注降解杂质的研究,杂质与药物临床使用的安全性密切相关,所以指定合理有效的药物杂质分析方法,控制药物中的降解杂质是一项非常重要的工作。首先要关注早期毒理样品的杂质研究,因为毒理样品的杂质水平是制剂限度制定的重要依据;其次关注原辅料的杂质,早期拿到原料后,尽可能早的将API与全辅料简单混合或模拟制剂工艺,制成剂型考察其在不同影响因素条件下的杂质产生情况;关注分析强降解实验产生的杂质,强降解实验又称破坏性试验,是在人为设定的特殊条件下,如强酸性、强碱性、氧化、高温、光照等引起药物的降解,可一定程度上了解该药品的内在稳定特性及其降解途径与降解产物;关注影响因素试验中产生的杂质,拿到原料后要尽早做预稳定性研究考察,对比原料和制剂的影响因素的稳定性差异,其对于保证临床用药安全起着非常重要的作用。
新药研发本身就很困难,而创新药研发更为难上加难,虽然创新药研发周期长、风险大,但是高风险对应的高收益,回报丰厚,怎样控制最少成本和最短的时间开发出适合临床所需的制剂产品,怎样尽早开发出和可上市的处方及工艺尽可能一致的产品是药学研发人员所必须具备的责任和能力,为此我们还是要树立正确的意识理念,药学研发人员的宗旨还是要为临床服务、为病人负责。
[1] 关于化学创新药临床试验期间药学变更的一些考虑
[5] 干货:API、制剂特性与溶出度、BE的相关性及案例分析PPT分享
本文来源于药品研发驿站















评论
加载更多