Nature:FDA或修改加速批准路径
时至今日,距离Aducanumab事件过去已将近一年。5月13日,《Nature》再次发文评论,这次争议性事件过后,FDA是否会修改“加速批准”(AA)路径?


今年5月,负责监督药物安全和生物医学研究的众议院能源和商业委员会(The House Committee on Energy and Commerce)宣布,如果一家制药公司未能在合理的时间内完成后续研究,希望能授予FDA更大的权力来撤销通过AA上市的药物。这项拟定中的条款,将作为FDA资金再授权法案(funding reauthorization bill)中的一部分,有望在今年9月前通过。
Aducanumab事件并不是AA遭受抨击的唯一原因。自AA启动以来,已有279种治疗药物通过AA路径上市,在过去的10年中,就有近三分之二的药物是通过该路径上市(图1)。华盛顿特区非营利组织国家健康研究中心(National Center for Health Research)主席Diana Zuckerman表示:“该路径变的越普遍,就意味着它的初衷已经改变。AA起初只是作为少数药物的特殊路径,但现在大多数抗肿瘤药物都通过AA或其他的加速审批路径。”
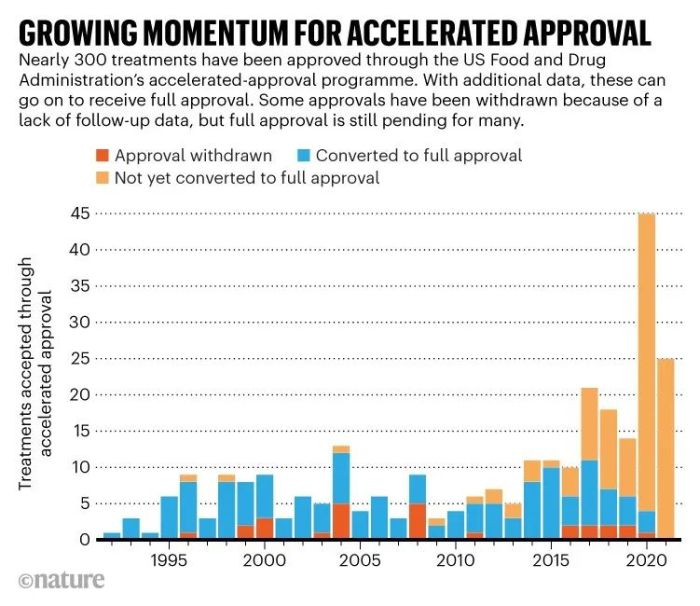
此外,要求进行后续研究是AA路径的一部分,但药企在进行后续研究方面的进展十分缓慢。但是,FDA强制要求他们提供数据的权力有限。此次立法提案或许能授予FDA更大的权力,尽管在众议院和参议院通过时仍可能发生重大变化。
今年2月,FDA局长Robert Califf在被任命前几天承诺,将把AA改革作为FDA的优先事项。接受《Nature》采访的研究人士也表示,为了保护该程序的完整性,当前急需改革,而立法提案是一个良好的开端。他们也建议加强机构监管和其他改革,以进一步防止制药公司滥用这条进入市场的途径。
华盛顿特区的非营利性组织“患者负担得起的药物”(Patients for Affordable drug)主席David Mitchell表示,制药公司没有按预期使用这项路径,反而有太多的企业设法找到这里面的漏洞然后加以利用。David Mitchell是FDA审查抗肿瘤药物的独立委员会的消费者代表。
被“速度”绑架的AA

FDA于1992年创立了AA路径,当时主要是为了应对HIV–AIDS危机,为了将急需的药物快速推向市场。基于这项路径,待审批的药物往往依赖替代终点指标,而不是像OS这样具有临床意义的终点,因为替代终点比传统临床试验终点能更快得出结果。
美国康涅狄格州耶鲁大学的流行病学家和全球健康专家Gregg Gonsalves是说服FDA通过这一路径的小组成员之一。他说:“我们推动这项加速审批的路径,是因为有人正在死去,我本人就是HIV阳性,我感到很绝望,需要获得希望。”
AA路径创立后,使得市场上的免疫疗法和新型抗肿瘤疗法的数量显著增加。其中一些药物临床获益的数据有限,却需要花费患者每年数十万美元。Gonsalves认为,该路径已被制药企业利用,用以加快审批的速度。与通过标准路径上市所需的时间相比,通过AA路径获批上市的抗肿瘤药物所需的时间可平均缩短3年,并可能用替代终点的单臂研究就能上市。
美国马里兰州约翰霍普金斯大学彭博公共卫生学院(Johns Hopkins Bloomberg School of Public Health)内科和流行病学家Caleb Alexander说,AA的一个重要问题是药企没有遵守他们的约定,及时对药物进行上市后研究以明确药物的临床获益。还有部分专家质疑,留给药企做确证性研究的时间是否太过宽松。2021年的一项分析发现,1992年至2016年间获得AA批准的药物中,有13%的药物在5年内没有转化为完全批准——未取得转化所需数据的情况下,它们在市场上的平均时间为9.5年。
罕见病公司联盟(Rare Disease Company Coalition)的一位发言人表示,药品上市后的临床试验可能需要很长的时间,尤其是针对进展缓慢的疾病,例如神经退行性疾病。此外,药企招募受试者也很困难,因为人们都想用经过完全批准的药物,而不愿意冒着服用安慰剂的风险。Diana Zuckerman表示,FDA并不能要求公司停止销售一种尚未转化为完全批准的药物,通常是公司自愿撤市。Caleb Alexander表示:一旦药品获得批准,FDA就会失去他巨大的影响力。
例如,2011年FDA认为贝伐珠单抗治疗乳腺癌缺乏临床获益,而撤销了其加速批准,这遭到了公众的反对,因为乳腺癌患者们希望能够维持批准。康涅狄格州丹伯里的非营利组织美国罕见病协会(National Organization For Rare Disorders)在2021年的一份报告中指出,对许多人来说,AA提供了“宝贵的希望”。但患有多发性骨髓瘤的Mitchell则认为:“给我希望的不是FDA,让我活下去的也不是希望,而是安全有效的药物。
FDA发言人Jeremy Kahn在一封电子邮件中表示,FDA致力于确保AA路径的完整性,对于患有严重疾病却缺乏治疗选择的人来说,他们愿意“接受临床获益存在一些不确定的新疗法”。他补充说,绝大多数AA药物的临床获益已得到验证。
改变法规就能解决问题吗?

FDA的AA路径已被其他国家的监管机构效仿,但欧盟和日本赋予他们的药品监管机构更多的权力,使其可以要求公司在规定的时间内提交确证性试验的数据,否则撤销其批准。
尚不清楚拟议中的法规变化究竟对FDA的监管会产生多大作用。这些条款会让FDA撤销批准更容易,但也可能延长撤销批准的官僚流程。Diana Zuckerman表示,她更倾向于坚持早期的提案,即一旦确证性试验逾期一年,就自动撤销批准。
Caleb Alexander则建议使用医疗保险作为杠杆。例如,位于巴尔的摩的美国医疗保险和医疗补助服务中心(CMS)可以决定对哪些药物提供资金。今年初,出于对Aducanumab疗效的担忧,CMS表示,将仅为入组临床试验的人群支付该药每年28800美元的费用。
这一决定几乎是史无前例的,Caleb Alexander认为,CMS应该考虑降低其他尚未获得完全批准的AA药物的报销比例。他说,这样的举动可以“给药企点一把火”,促使他们完成试验。“当我们不知道某种药品完整的安全性和有效性,为什么纳税人要为它全部的价格埋单呢?”他这样问道。
但Mitchell担心,削减报销比例会让药企失去研发临床急需药物的动力,在确诊骨髓瘤后,他觉得是3种加速审批的药物让他活下来了。他还表示Aducanumab事件只是在AA这一伟大程序中的一次“波动”,这使得人们更多的关注到需要对一些药物进行验证性试验。改革不是那么简单的,一旦一种药物进入市场,药企并不急于找一个理由让它停产。
尽管如此,许多研究人员和药物安全支持者仍渴望看到变革。Diana Zuckerman说:“我们开始试图修正一个钟摆,它往一个方向上走偏了,看看现在吧,我们在这个方向上走了多远”
本文来源于医药魔方
邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多