关于美国药品GMP法规框架,你了解多少?
一起来看一下:

关于 21 CFR

CFR,Code of Federal Regulations,美国联邦法规。
CFR的TITLE 21,也就是我们说的 21 CFR,是关于食品和药品的联邦法规。
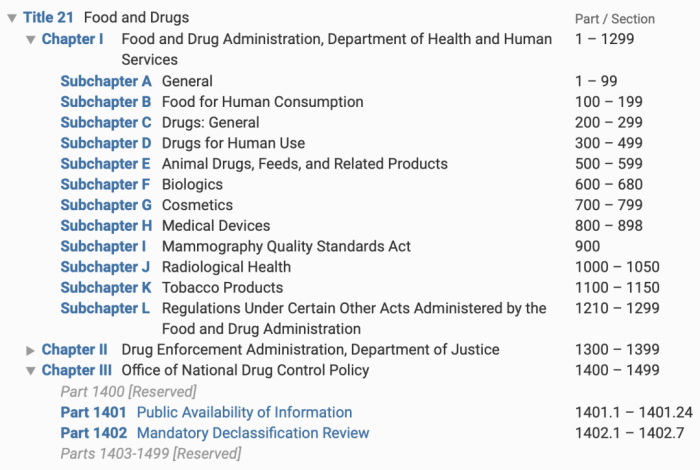
而21 CFR中,共有三个章节,分别是:
-
第I章:食品和药品的监管,人类健康服务部门 -
第II章:药品的强制监管,执法部门 -
第III章:国家药品控制政策办公室
我们所知道的《Part 11:电子记录和电子签名》,在第I章下的子章节A Subchapter A,通则General。
子章节A下共有99个部分(Part),电子记录和电子签名是Part 11,而编号为Part 83-98是预留的。

而Part 210和Part 211,则是在第I章下的子章节C (Subchapter C), 药品通则(Drugs general)中。在子章节C,目前目前共有19个Part,当然,编号从200开始,也是编到了299。


关于 Part 210
Part 210中共包括3个小节,分别是:

210.1 cGMP法规的状况
210.2 cGMP法规的适用性
210.3 定义
临床研究用药品,要符合子章节D部分Part 312的要求(也就是不用按Part 210和211来管理)。
同样,欧盟GMP的附录13,也是关于临床研究用药品的,当前生效版是2010年版,不过,在2017年也提出了修订版意见,要将临床研究用药品的GMP与上市药品的GMP的一些具体要求加以区分。这是闲话了。

关于 Part 211
Part 211是药品制剂的cGMP。它的结构分为从A-K共11个部分。
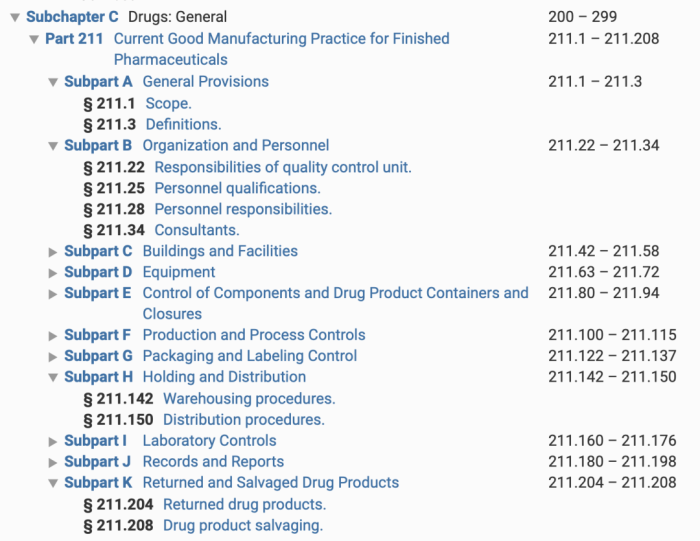
这11个部分,分别是:
A:基本条款
B:机构与人员
C:厂房设施
D:设备
E:组分、药品容器和包装的控制
F:生产和过程控制
G:包装和贴签控制
H:保存和分配
I:实验室控制
J:记录和报告
K:退货和收回
这个结构似曾相识,但是,又与中国GMP或者欧盟GMP很不相同,是不是?我们分别来看一下其中的各个章节。章节中的内容与中国GMP和欧盟GMP明显不同的,我们标黄来显示:
A章节,基本条款。相当于通则。下面包括范围和定义;

B章节:机构与人员,这个框架我们并不陌生。下面细分为4个小节:质量控制单位的职责、人员资质、人员职责、顾问。“顾问”在中国GMP中是没有的,但在欧盟GMP和美国GMP中,以及ICH Q7中,都有这个专门的小节。并且,许多FDA的警告信中,都会专门提到:你们可以聘请专门的顾问来帮你们改善GMP方面的问题。关于顾问:顾问有什么用?!—— ICH Q7,认知,解读与实践:11

C章节:厂房设施。下面分为:建造和设计细节、照明、通风/空气过滤/暖通、管道系统、排污排废、清洗和盥洗、卫生、维护共七个小节。

D章节:设备。下面分为:设备设计/尺寸和位置、设备构建、设备清洁和维护、自动化/机械化和电子设备、过滤器共5个小节。

E章节,我们可以理解为《物料和产品》,相当于中国GMP的第6章(而欧盟GMP则把物料和产品的控制放在《生产管理》中了);这个章节下面有:基本要求、未检物料的接收与贮存、物料的检验和批准或否决、批准物料的使用、批准物料的复验、物料否决、药品容器和密封共7个小节。

F章节和G章节(生产和过程控制+包装和贴签),我们可以理解为《生产管理》,相当于中国GMP的第9章。
F章节下分为8个小节,分别是:书面程序及偏差、投料、收率计算、设备标识、过程物料和药品的取样和检验、生产时限、微生物污染控制、返工。
G章节下分为6个小节,分别是:物料(包装)的检查和使用标准、标签发放、包装和贴签操作、OTC人用药品的防伪包装要求、药品检查、有效期。

H章节:保存和分配,我们可以理解为中国GMP的《产品发运和召回》(但是不含召回的内容,召回不在Part 210和211中)。这下面分为2个小节,分别是:仓贮程序、分配程序。

I章节:实验室控制,相当于中国GMP中的第十章中质量控制的章节吧。下面分为7个小节,分别是:基本要求、检验和放行、稳定性检测、特殊检验要求、留样、实验室动物、盘尼西林污染。

J章节:记录和报告,类似于中国GMP中的第八章中文件管理的部分。下面分为9个小节,分别是:基本要求、设备清洁和使用日志、物料成分/包材和密封及标签的记录、主生产记录和主控制记录、批生产和批控制记录、生产记录审核、实验室记录、分配记录、投诉文件。

K章节:退货和收回药品。退货的部分,在中国GMP中是分布在几个不同的章节的,而在欧盟GMP中,是在第八章。

收回药品,在中国GMP和欧盟GMP中都未提及,只有在美国GMP中,在Part 211中,有专门的章节去谈收回药品。怎么理解药品收回呢?退货(无论是否有质量问题)、召回(基本上都是有大的质量问题),当回到厂里后,都是一种收回。
在Part 211中强调:经过不正常的贮存条件的药品,包括严苛的温度、湿度、烟雾、压力、时间或由于自然灾害导致的辐射、火灾、事故、设备失败等,都不应当收回并重返市场。如果怀疑药品曾经经受过这些条件,在经过一系列的检测调查证明后,才可以考虑收回。


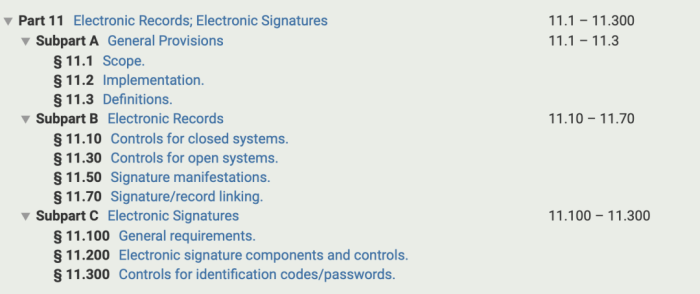
Part 11 电子记录和电子签名
-
A:基本条款。包括范围、实施、定义; -
B:电子记录。包括:闭式系统的控制、开放系统的控制、签名表征、签名/记录的链接; -
C:电子签名。包括:基本要求、电子签名的组成和控制、识别码/密码的控制。

写在最后
中国现行GMP是以欧盟GMP为基础来建立的。而对于美国的GMP来说,虽然基本的方向与欧盟和中国类似,但是无论是框架,还是条款和一些细节要求,都有较大的差异。可以作为我们在思考和理解某个部分的GMP活动的要求时的补充。
对于原料药,欧盟和美国都采用ICH Q7来作为GMP要求的指南。中国GMP专门编制了《附录2原料药》,却是一个“缩减”版的ICH Q7。
对于无菌药品的GMP特定要求,欧盟和中国,都作为了附录一。而美国,则有一个2004年的以无菌制备来生产无菌药品的GMP指南:

邵丽竹
何发
热点文章
-

重磅,新版GCP发布,9月1日起施行!
2026-06-08
-

无菌药品生产环境监测性能确认的研究及应用
2026-05-07
-

中药提取自动投料系统(模式)设计与应用——以华润三九和华润江中投料系统为例
2026-05-20
-

2025年度中国医药工业主营业务收入前100位企业发布!哪家企业上榜?
2026-07-13
-

预灌封注射剂生产工艺管理要点概述
2026-05-12
-

CDMO龙头三星生物罢工!中国CDMO企业迎订单转移窗口期?
2026-05-12
-

论医药洁净区空间消毒 / 灭菌的常用方法
2026-06-26
-

基于CFD仿真技术的灌装机充氮装置设计优化
本文以某制药产线的灌装机设备为研究对象,采用计算流体动力学(CFD)仿真技术对充氮装置的充氮性能进行分析,并结合分析结果对氮幕结构进行了优化设计。随后,针对优化方案进行性能仿真验证,结果显示优化后的顶空残氧量降低至0.252%。为了进一步验证优化方案的实际效果,将优化方案应用于实际产线进行性能测试,测得的顶空残氧量为0.68%,这一结果满足了小于1%的要求,表明其充氮保护性能已达到国际先进水平。
作者:
-
药品密封性检测 :用户需求与优化
-
可控冻融系统在生物原液上的应用
-
人用疫苗生产数字化转型
-
药包材生产质量管理的进阶策略
-
药厂洁净区域风量和压差的控制策略





评论
加载更多