
药企如何合规地应用一次性生产工艺和使用一次性设备
随着国内生物医药产业快速发展——各家药企为加快申报速度、节省固定资产成本,一次性工艺成为越来越普遍的选择。
国内生物医药企业竞争焦点相对集中,对速度要求更高。国内市场相对欧美市场较小,1000 L、2000 L通常可以满足国内市场的需求。这也导致了未来相当一段时间内,使用一次性反应器和一次性生产工艺会更加普及。
一次性工艺具有固定资产成本低、建设周期短等优势,但也面临缺乏标准化、溶出/析出验证等挑战。
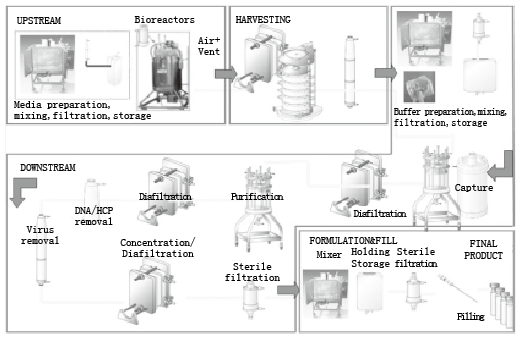
图1 生物工艺流程
溶出物/析出物简介
溶出物(Extractables):为了评估工艺风险、选择合适的工艺材料,在比工艺条件更剧烈的,如酸性、碱性、有机相等条件下,与产品接触的材料迁移到溶剂中的化合物。因此也可以认作“合理最差条件溶出物”。与析出物相比,更侧重一次性工艺材料的检测,通常由一次性产品供应商提供。
析出物(Leachables):在正常的工艺条件下,与产品接触的材料迁移到溶剂中的化合物。析出物(Leachables)通常为溶出物(Extractables)的子集。与溶出物相比,更侧重在特定药物生产过程中的检测,通常由一次性产品使用者提供,并通常需要供应商的协助。

图2 溶出物与析出物之间的关系
由于析出物(Leachables)不一定可以通过后续工艺去除,可能存在于最终产品中,从而对药品安全性和有效性产生影响,因此检测析出物非常重要。
实际上由于析出物含量一般很低,且可能和活性组分反应形成次级析出物,有时候需要经过长期稳定性研究才可能检测到某些析出物,或者无法准备鉴定和检测。
实际应用中,通常是先检测溶出物(Extractables),即合理最差条件的溶出,然后用溶出物评估析出物水平,并根据用药剂量评估安全性,如果存在潜在风险,再在此基础上进行析出物研究。
溶出物(Extractables)和析出物(Leachables)包括:
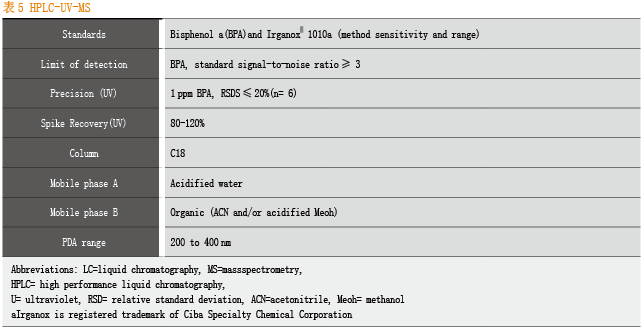
挥发性有机物:通常采用Headspace GC-MS分析;
半挥发性有机物:通常采用GC-MS或者(高分辨率质谱HRAM)GC-MS分析;
非挥发性有机物:通常采用(高分辨率质谱HRAM)LC-MS/MS分析;
微量元素(金属):通常采用ICP-MS分析。
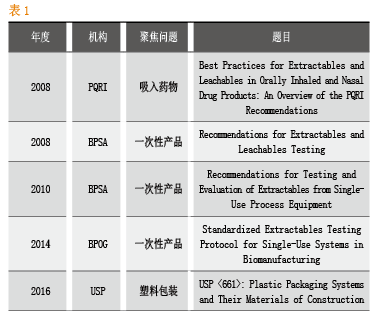
溶出物/析出物研究的标准化
截止到目前,工业界没有标准化的溶出物和析出物的检测方法,但面对一次性工艺的日益普及,监管部门和业界都在努力寻求建立相关标准。聚焦生物药物一次性生产工艺的主要包括BPOG、BPSA等行业协会。
BPSA全称BioProcess Systems Alliance,是26家一次性工艺产品的供应商联盟,成立于2006年。
BPOG全称Biophorum Operations Group,是由超过53家生物医药企业和工艺产品供应商等组成的行业协会。
BPOG针对一次性工艺的标准化做出了持续努力,2014年推出的Standardized Extractables Testing Protocol for Single-Use Systems in Biomanufacturing,对一次性产品溶出测试的标准化提出了完整Protocol,由于该方案同时吸取了药企和一次性产品供应商以及监管部门的意见,因而具有很好的参考价值。



BPOG溶出物研究标准化Protocol
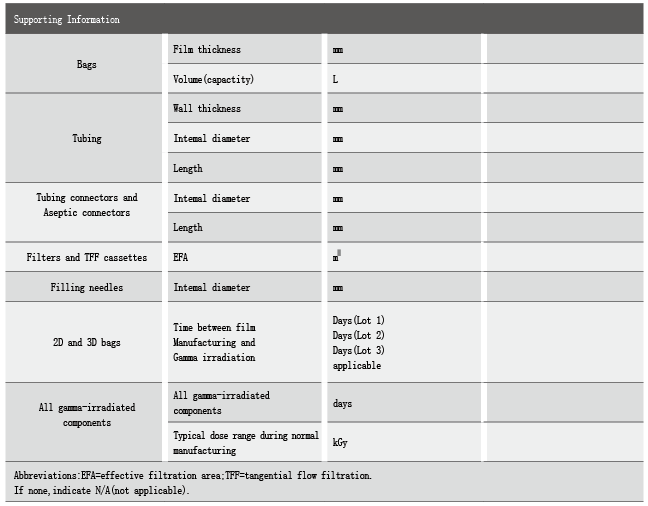
BPOG溶出物研究方案对试验预设条件、试验具体条件(不同产品对应的暴露温度、暴露时间)、溶出物研究报告都给出了具体的建议。
尽管生物药最长责任人仍为药企本身,但该方案对于一次性产品供应商的溶出物研究提出了规范、详细的要求,为药企顺利申报,节省验证时间提供了保证。
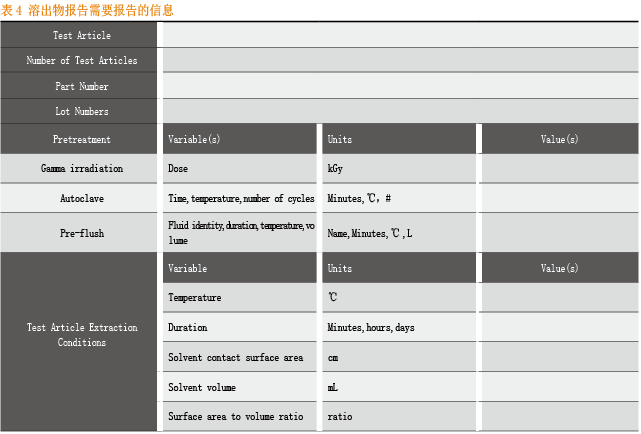
对于不同异地性产品,BPOG给出了溶出物研究的具体记录信息和预设条件。
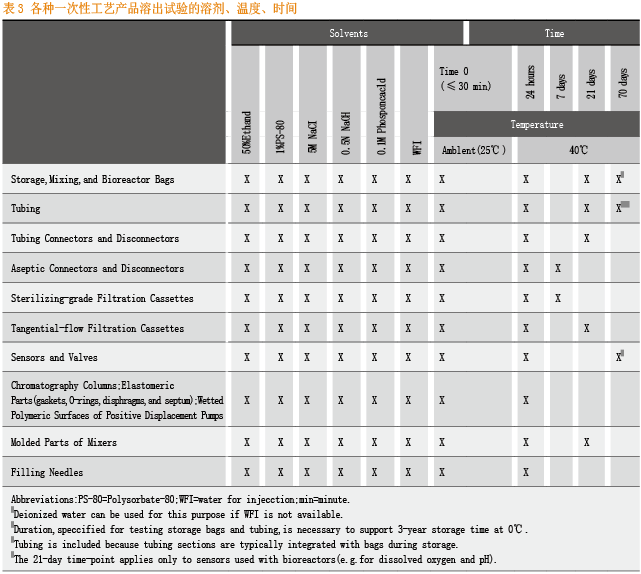
对于不同一次性工艺产品的溶出试验,BPOG给出了不同溶剂、暴露温度、暴露时间的量表。
对于溶出物研究报告,则应该包括以下几个部分:
溶出物研究综述;
溶出试验预设条件的详细介绍;
一次性袋子生产;
gamma射线灭菌;
溶出物验证的时间间隔;
一次性袋子/管路的厚度;
液体接触材料的材质信息;
微量化合物信息等。
对于常用的分析方法,BPOG也给出了标准化的参数。
总体来看,BPOG的溶出物研究方案非常具体而详细,同时由于吸收了很多药企和一次性产品供应商的意见,对于整个生物制药行业都具有非常重要的意义。
药企如何合规地应用一次性工艺确定
BPOG的溶出物研究Protocol可以规范一次性产品供应商的产品合规与形式合规,但对于作为药品质量最终责任人的药企,到底如何合规地应用一次性产品呢?
BPOG在“生物制药一次性聚合物产品析出物风险巩固指南”中给出了操作性较强的流程。
溶出物/析出物研究的内容和深入程度,应该依药品研发阶段的变化和一次性产品的材质风险高低而定。
在前期研发和I期、II期临床阶段,应该搜集一次性产品的材质、来源、储存/运输条件、生产商信息、包装信息、生产条件理化相容性、USP Class IV/EP3.1.9法规相容性、溶出Profile等基本信息,当药品研发进入三期临床阶段,应进行相对系统的溶出物/析出物评估,包括溶出物Profile、风险评估、毒理评估、析出物研究/报告等。
在溶出物/析出物研究过程中,如何进行有效的风险评估非常重要。BPOG在指南中给出了较为详尽并具操作性的模型。
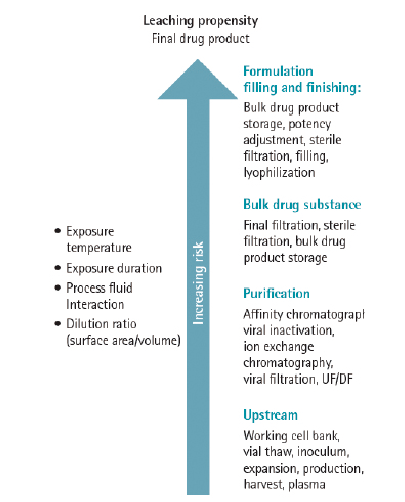
对于一次性产品而言,工艺流程距离和工艺条件苛刻程度直接关系到对与终产品的质量,越下游的工艺环节风险越大,越苛刻的工艺条件风险越大。
因此,对工艺流程距离和工艺条件进行风险等级的确定和权重的打分,可以对风险分值进行量化,进而采取相应的进一步研究方案。


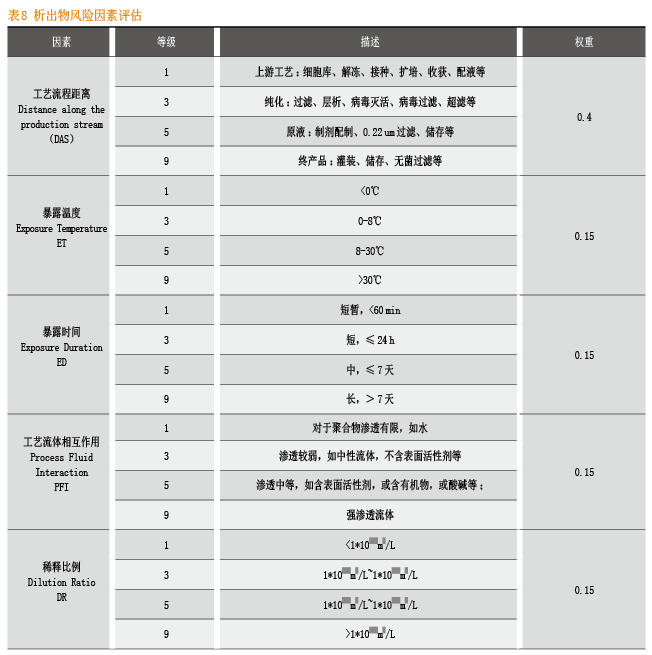
根据上述的风险等级和权重,可以按照以下公式计算析出物总风险分值:
(2000 L Bioreactor, post bioreactor to cell remove process, use 3/4" ID Advantaflex TPE 2meters tubing to transfer,transfer time is around 65 mins.)
UPSTREAM(1*0.4)+ Temperature 8-30℃(5*0.15)+transfer time is around 65 mins(3*0.15)+ Process Fluid Interaction(3*0.15)+ Dilution Rtio(1*0.15) = 2.2
LRR=2.2 低风险
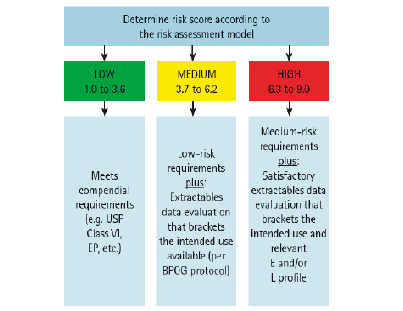
根据LRR的范围,可以确定析出物风险高低:
* LRR=6.3~9.0,高风险;
* LRR=3.7~6.2,中风险;
* LRR=1.0~3.6,低风险。
根据不同风险等级,可以采取相应的研究方案:低风险可以认为满足USP Class IV和EP相关法法规要求;中风险应该按照BPOG的溶出物Protocol进行溶出物数据评估,或者药企In-House的析出物研究,以证明风险可控;高风险应该在中风险措施基础上,进行工艺特异性的析出物研究。
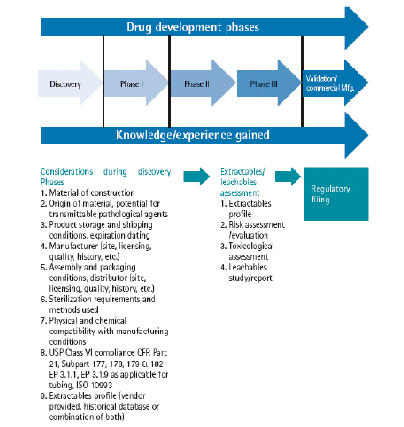
此外,如果高风险情况下,基于溶出物Profile的毒理评估,发现每剂量暴露量超出安全范围,则需要进行相应的析出物研究以证明风险可控。注意,进行这类析出物研究的必要性,仅在供应商无法提供相应的溶出物数据,或者其数据不符合当前产品的生产工艺条件时。
综上,药企可以根据药品生产工艺,进行特定类型一次性产品的风险评估,或者转而调整工艺,降低产品质量风险。既有操作意义,也有一次性工艺指导意义。
在上游工艺和温和条件工艺中,可以更加方便评估并合规地使用一次性产品;对于下游工艺和苛刻条件工艺,也可以有效评估风险,以决定是否采用一次性产品,或者如何选用不同供应商的一次性产品。
BPOG析出物风险评估模型,对于药企如何合规地应用一次性产品有极大的现实意义。一方面方便药企进行一次性工艺的风险评估,另一方面,标准化的风险评估模型也便于监管部门进行审评,有利于促进整个行业内的一次性工艺普及。
(本文由上海亮黑科技有限公司提供)


![]()
2001-2009Vogel Industry Media版权所有 京ICP备12020067号-15 京公网安备110102001177号


加载更多