
一次性系统溶出物/析出物验证方法
在制药领域,采用一次性系统(Single Use Technology,SUT)有助于快速建立生产线、节约投资成本、灵活调整工艺以适用于不同产品的生产,同时可以避免交叉污染、减少清洗投入,为终端用户提供了很大便利,因此一次性系统在制药工业中的应用日趋广泛[1]。出于药品质量、效能和安全的考虑,对可能引入药物制剂中的杂质进行研究不仅非常重要,而且十分必要。对此,国内外的法规和行业机构都有类似要求[2,3]:“…与料液接触的表面不得因与产品发生反应、释放物质或吸附作用而对产品质量造成不利影响…”[4],溶出物(Extractables)/析出物(Leachables)验证可以确认可能迁移至药物制剂中的化合物,用以评估一次性系统对药品质量、效能和安全的影响。
明确溶出物和析出物的定义以及两者的区别非常重要,以避免混淆。溶出物指的是在夸大的时间和温度下,从任何与产品接触的材料迁移到合适溶剂中的化合物,代表了绝大多数可能的析出物[5],其来源可能为过滤器或一次性系统材质中聚合物的单体、寡聚物、降解产物、相关添加剂(如抗氧化剂、润滑剂、稳定剂、增塑剂、改性剂等加工助剂)以及生产过程中的溶剂残留等等。析出物是指在实际的工艺条件下,从任何直接与产品接触的材料,包括弹性体、塑料、玻璃、不锈钢或者涂层材料迁移进入药物制剂的化合物,并可能存在于最终药品中,直接反映对药品可能产生的影响,通常是溶出物的一个子集[5]。对于一次性系统来说,评估“工艺析出物”的含量是很重要的,“工艺析出物”是指那些从接触设备迁移进入生产工艺线的化合物,不一定能通过后续的工艺步骤去除,比如层析、切向流或丢弃起始产品。当夸大条件下的溶出物研究结果表明一次性系统对药品质量产生干扰时,需要考虑进行析出物研究。
为获得产品中的析出物数据,使用药物产品直接进行析出物研究似乎是最直观的做法。然而药物产品中往往含有高浓度非挥发性物质,且成分复杂。相对而言,析出物的浓度很低,其分析会受到药物组分的干扰(比如掩盖析出物),从而造成不准确、甚至假阴性的结果。因此,目前公认比较科学的做法是:首先检测合理最差条件下的溶出物数据,用溶出物数据评估析出物水平,然后基于药物每人每日摄入剂量进行安全评估;如果评估结果显示有潜在风险,再在溶出物的基础上进行析出物研究[5]。
与药物产品兼容并直接接触药物的过滤器或一次性系统,影响其溶出物 / 析出物的因素有两类:一类为工艺条件,如时间、温度、灭菌条件和接触面积;另一类为产品特性,如产品中的有机溶剂、表面活性剂、产品pH值等。
溶出物验证试验可分为两步进行:首先根据产品特性和工艺条件,确定试验流体(如模型溶剂)和试验条件(如温度、时间、料液pH值、接触面积、预处理步骤等),与过滤器或一次性系统充分接触(如循环、静态浸泡、振摇等),获得溶出物样品;然后对样品作必要的预处理,再进行定性和定量分析,检测其中化合物的种类和含量[6,7,8]。
析出物试验跟溶出物试验步骤类似,主要有两点区别,一是采用产品溶液作为试验流体,二是采用实际工艺参数作为试验参数。如果产品溶液中的组分比较复杂,定性和定量分析前的预处理方法可能也跟溶出物的预处理方法不一样。
溶出物/析出物的分析方法
分析方法选择
对于单个一次性组件或简单的一次性系统,溶出物/析出物的提取方法相对比较容易,而对于溶出物/析出物分析,其方法的开发需要更多资源。随着分析技术的快速发展,对溶出物/析出物的分析检测也日趋深入。由于溶出物/析出物的组分十分复杂,单一的分析方法难以检测所有的目标化合物。国内外各行业机构推荐的方法主要包括:液相色谱质谱联用分析法(Liquid Chromatography/Photodiode Array/Mass Spectrometry,LC/PDA/MS)、气相色谱质谱联用分析法(Gas Chromatography/Mass Spectrometry,GC/MS)、电感耦合等离子体质谱法(Inductively Coupled Plasma/Mass Spectrometry,ICP/MS)、离子色谱法(Ion Chromatography,IC)、紫外光谱法 (Ultraviolet Spectroscopy, UV)、傅里叶红外光谱法(Fourier Transform Infrared Spectroscopy,FTIR)、核磁共振波谱分析(Nuclear Magnetic Resonance,NMR)、非挥发性残留物(Non-volatile Residue,NVR)、总有机碳(Total Organic Carbon,TOC)、pH 值、电导率等等[8,9]。其中 LC/MS、GC/MS、ICP/MS 和IC均可用于定性和定量分析,UV、FTIR、NMR 主要用于定性分析,NVR 用于常规定量分析,TOC、pH和电导率则作为辅助分析。
液相色谱质谱联用分析(LC/PDA/MS)
液相色谱(LC)主要进行溶出物 / 析出物的色谱分离,通过二极管阵列检测器(Photodiode Array,PDA)和质谱检测器(Mass Spectrometry,MS)检测一定波长和质荷比(m/z)范围内的非挥发性和部分半挥发性化合物。二极管阵列检测器检测可获得具有紫外吸收的化合物信息,质谱检测则可获得化合物的分子量及结构信息,与标准品的保留时间(Retention Time,RT)和质谱匹配进行化合物的鉴别,并根据标准曲线对化合物定量。使用大气压化学电离(Atmosphere Pressure Chemical Ionization,APCI)和电喷雾(Electrospray Ionization,ESI)两种离子源方式,可互相补充,更全面的分析溶出物/析出物。对于高盐、高 pH、低 pH、含表面活性剂或含蛋白质的样品,进行液相色谱分析前需对样品预处理[11]。
气相色谱质谱联用分析(GC/MS)
质谱检测器(MS)与气相色谱(GC)联用,通过顶空进样气相色谱质谱联用(Headspace-GC/MS)和直接进样气相色谱质谱联用(Direct Injection-GC/MS)两种不同的进样方式分别检测挥发性和半挥发性化合物。对于一次性系统的溶出/析出样品分析,采用两种GC/MS方法分别进样分析,可以更加全面检测出样品中的挥发性和半挥发性物质。溶出或析出样品可直接通过顶空进样 GC/MS进行分析,检测其中所含的挥发性化合物。通常,这些化合物产生于伽马灭菌过程,而且可能随着放置时间增加而减少。因此必须在灭菌结束后尽快分析。挥发性化合物在较早取样点的样品中通常相对较多。直接进样GC/MS能够检测到一系列半挥发性化合物。一些溶出或析出样品能够直接用GC/MS色谱柱进样分析,然而对大多数水溶液样品,分析其中的半挥发性化合物需要进行液液萃取,比如使用与色谱柱和分析方法兼容的二氯甲烷溶剂进行萃取,然后再进行分析。
通过GC/MS图谱获得化合物的分子量及结构信息后,对照标准品的保留时间和质谱匹配对化合物进行鉴别,并利用化合物响应(通常是峰面积)对比标准曲线进行定量。必要时可采用其他检测器,如氮磷检测器、氢火焰离子化检测器等。对于高盐、含表面活性剂或含蛋白质以及极端pH值的样品,进行直接进样气相色谱分析前通常需对样品预处理,进行顶空进样色谱分析的样品则一般无此要求[11]。
电感耦合等离子体质谱法(ICP/MS)/电感耦合等离子体发射光谱法(ICP/OES)
这是一种用来测定元素含量的方法,具有灵敏度高、速度快、检测限低等优点。其原理是被测元素通过一定形式进入高频等离子体电离成离子,产生的离子经过离子光学透镜聚焦后进入四极杆质谱检测器,按照荷质比进行分离。适用于环境、地球化学、半导体、核工业、食品药品、生物制品等不同领域几乎所有金属元素含量的分析,也可以分析绝大部分非金属元素。此外,如果能够达到符合要求的专属性及检测限值,电感耦合等离子体发射光谱(ICP/OES)作为另一种检测方法,同样可用于检测元素含量[11, 12, 13]。
离子色谱(IC)
离子色谱是利用被测物质的离子特性进行分离和检测的一种液相色谱法,主要用于测定各种离子的含量。 根据分离原理的不同,可分为离子交换色谱、离子排斥色谱和离子对色谱,其中以离子交换色谱最为常用。离子交换色谱采用低容量离子交换树脂为固定相进行分离,通过电导检测器连续检测流出物电导变化来分析阳离子和阴离子。除电导检测器外,离子色谱还有直流安培、脉冲安培和积分安培等电化学检测器以及紫外-可见和荧光检测器。
在 SUT 的溶出物和析出物分析中,各种分析方法的互补特性几乎可以涵盖绝大多数化合物的分析,因此 IC 应用较少。
紫外光谱法(UV)
紫外光谱法(UV)可分析溶出物/析出物中带有紫外发色基团的化合物,通常为含不饱和化学键和含孤电子对的化合物。有些样品中的溶质或溶剂在特定波长下也具有较强的紫外吸收信号,可能会覆盖溶出物/析出物的吸收峰。分析溶出物/析出物时,采用LC/PDA/MS分析方法能够覆盖紫外光谱法,因此可不单独进行紫外检测。
非挥发性残留物(NVR)
非挥发性残留物(NVR)是一种定量评估溶出物的分析方法。作为试验流体的挥发性模型溶剂蒸发后,将非挥发性溶出物烘干所获得的物质称为非挥发性残留物,通过称重的方法来确定最差条件下从药品生产设备迁移到模型溶剂中的非挥发性残留物的量,从而代表在工艺条件下可能从生产设备迁移进入到药物产品中的非挥发性残留物的最大可能数量。非挥发性残留物通常来源于高分子材质的寡聚物、添加剂及降解产物等。由于药物产品中往往也含有非挥发性组分,干扰非挥发性残留物总量分析,因此该方法不适用于析出物分析。
傅里叶红外光谱法(FTIR)
红外光谱法(FTIR)是一种强有力的基本定性分析工具,通过与特定的图谱匹配来表征功能基团以及化合物种类。将红外光谱图与相同化合物进行比较,可推断出溶出物中部分非挥发性残留物(NVR)的种类信息。然而,为获得单一化合物的定性与定量信息,则必须借助LC/PDA/MS 或GC/MS的分析方法才能实现。
总有机碳(TOC)、pH 值、电导率
总有机碳(TOC)的测定是一项非常重要的分析方法,能够直接、快速、真实、灵敏地反映溶出物水平,然而,其局限在于仅适用于电导率相对较低的无机溶液。因此,常与pH值、电导率共同作为三项常规的辅助分析方法,用于考察一次性系统与试验流体接触前后的变化情况,这些参数的变化通常是由溶出物/析出物所造成,因此这些数值对评估一次性系统是否影响药物产品有一定帮助。
分析方法开发
为了保证分析方法的可靠性,以准确定性和定量溶出物/析出物,应当预先对分析方法进行开发和验证,同时应设立一定的接受标准[10]。药品生产设备的供应商,应该建立灵敏的色谱方法,从而更好鉴别可能引入药品生产工艺线中的聚合物、聚合物中的添加剂以及聚合物相关的降解产物。通常一次性系统供应商更能理解、控制并鉴别一次性系统生产时所使用的聚合材料。下文以LC/MS和GC/MS为例,简单介绍分析方法的开发和验证。
专属性(Specificity)
专属性是指在其他成分可能存在时,采用的方法能正确测定出被测物质的特性。一次性系统的供应商难以全面控制所有生产一次性系统的原材料信息,比如树脂生产商没有公开的配方或是一次性系统中所用的半成品材料。然而,有的供应商已经较好地建立了源于一次性系统材料并可能出现在药品中的化合物的保留时间和标准品谱库。在分析溶出物/析出物时,上述标准品库中的特定标准品与被测物质在相同情况下进样分析,识别被测物质,可以获得很高的置信水平,从而验证其专属性。如果仅仅通过出版的化合物数据或谱库提供的信息与被测物质的保留时间或质谱片段进行比较,不能准确识别被测物质。
灵敏度(Sensitivity)
灵敏度通常以检测限(Limit of Detection,LOD)和定量限(Limit of Qualification,LOQ)来表征。检测限(LOD)指样品中被测物质能被检出的最低量,定量限(LOQ)指样品中被测物质可被准确定量的最低量。
信噪比(Signal to noise, S/N)法常常被采用来测定灵敏度。比较已知浓度标准品测出的信号与空白样品测出的信号,分别将信噪比为3:1(S/N=3:1)和10:1(S/N=10:1)时的相应浓度确定为 LOD和LOQ。不同的一次性系统组件,其溶出物 / 析出物不尽相同,宜选择具有代表性的化合物作为标准品,获得其LOD和LOQ值以表征灵敏度。分析溶出物 / 析出物时,对于某个具体方法, 报告其通用的LOD和LOQ值(比如低于0.1 ppm)是不恰当的,因为化合物类型和基质不同,其响应因子和离子化效能会发生变化,因此,应当报告该类化合物具体的LOD和LOQ值。
准确度(Accuracy)
准确度是测定结果与真实值的接近程度,一般用实际测得的数值与配制的真实浓度值的百分比(%)表示,比如85%。将标准品直接加入基质中计算出该百分比以考察方法的准确度,其结果应符合一定的接受标准。
分析溶出物时,一般选用一次性系统中容易或经常迁移出来、并且在相应分析方法中具有代表性的化合物作为标准品来考察准确度;分析析出物时,准确度则通过溶出物试验中识别出的化合物作为标准品来衡量。不论分析溶出物或者析出物,进样分析前常常需要对样品进行预处理。这是因为有的溶剂(如水)不适合直接进样气相色谱,有的药物组分(如蛋白质或表面活性剂)会污染或堵塞色谱柱。预处理虽然费时,但是可以获得具有代表性的分析数据并避免伤害分析仪器。常见的预处理方法包括液液萃取、固相萃取、蛋白沉降等方法,通过考察所添加的标准品的回收率来确认预处理方法是否恰当并且帮助评估溶出物及析出物的量。由于模型溶剂组分相对简单,对于溶出物样品较易确定预处理方法。这有利于日后确定析出物样品的预处理方法,并且由溶出物数据提供佐证,可以更准确地识别析出物中含量较低的化合物,避免漏检化学物质或造成假阴性结果,影响后续安全评估的客观性。
对比产品对照和析出物的谱图(图1和图2),很容易识别出部分析出物,如位于RT=0.52 min, 2.27 min和3.24 min处的吸收峰。产品对照的谱图中,RT=1.65 ~ 2.00 min处有一个很强的吸收峰,不能确定该处是否存在析出物。对比溶出物谱图(图3),RT=1.89 min处存在吸收峰,是潜在的析出物,对析出物进一步进行离子扫描分析(图4),确认该物质属于析出物。再对该物质进行定性和定量分析,得到准确的分析结果。
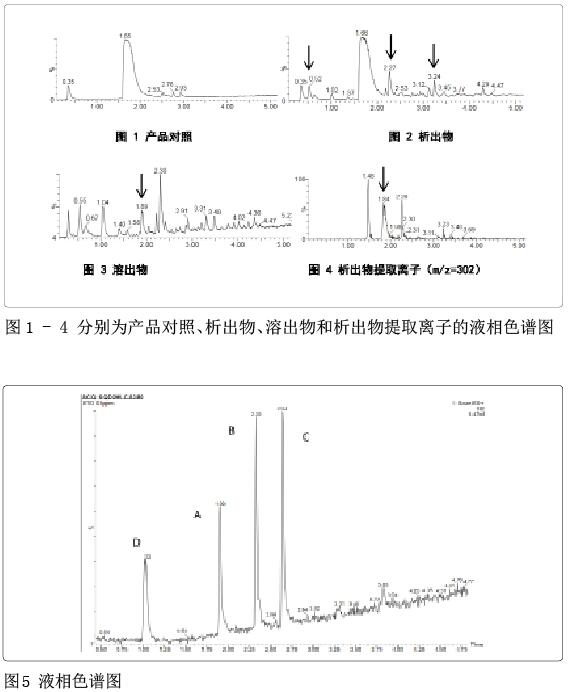
可见,溶出物数据有利于发现潜在的析出物,可以最大程度地识别出析出物种类,避免析出物漏检或假阴性。同时在极少数情况下,析出物抽提液中可能还含有迁移出的化学物质与药物组分反应产生的微量反应产物。以溶出物谱图作为参照,可以识别出可能的额外反应产物,从而得到更准确的析出物数据,使后续的安全评估更加客观真实,降低风险。
精密度(Precision)
精密度是指在规定的测试条件下,同一个被测样品经多次取样测定所得结果之间的接近程度,一般用偏差、标准偏差或相对标准偏差表示。
相同条件下,同一个分析人员测定结果的精密度称为重复性。同一个实验室,分析时间、分析人员和分析设备这三个因素(甚至包括试剂、温度、进样方法等可能引起试验结果偏差的所有因素)当中,变动其中任何因素,其测定结果之间的精密度称为中间精密度。不同实验室、不同分析人员之间测定结果之间的精密度称为重现性。
样品分析前,须考察系统适用性,如检测限、定量限、精密度、准确度等等,以确保仪器运行正常、状态良好、结果稳定可靠。作为系统适用性的考察项目之一,每次分析溶出物/ 析出物样品前,用标准品溶液连续多次进样,根据保留时间和峰面积分别确定其相对标准偏差,并且在样品运行过程中用已知浓度的标准品溶液考察其响应值的偏差,以确认分析结果的有效性。
线性和范围(Linearity and Range)
线性是指在一定范围内,测试结果与被测物质浓度直接呈正比关系的程度。范围是指能达到一定精密度、准确度和线性,测试方法使用的高低限浓度的区间。通常标准曲线应当包含至少5个浓度点,并保证样品浓度落在标准曲线范围内,最低浓度点为定量限浓度水平。通常,如果使用相关的一类物质时,半定量分析是一种可以接受的方法。其优点在于可在较短时间内消耗较少的资源,获得化合物大致的浓度结果。此外,当不能通过商业途径获得标准品时,采用半定量分析不失为一个比较合理的选择。如果需要更加准确的结果评估高风险工艺,则需要将标准品加入基质当中,获得线性范围内不同浓度的标准曲线,从而进行定量分析。
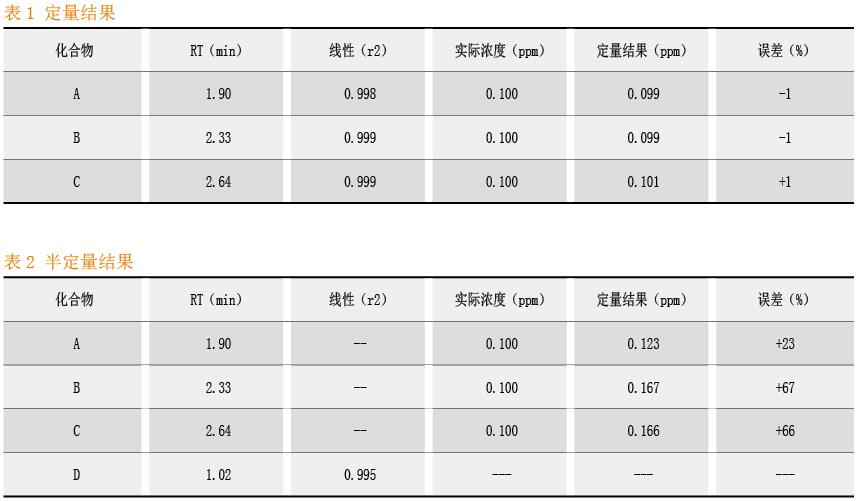
由于不同化合物在仪器中的响应因子(即峰面积与浓度的比值)不同,半定量分析不能确定化合物的准确浓度。如图5所示,液相色谱图中3个吸收峰A、B、C分别代表3个不同的化合物。分别采用A、B、C的标准品进行定量分析以及采用另一个化合物D对其进行半定量分析,两种定量结果见表1和表2。在多数情况下,定量分析比半定量分析的结果更准确。从结果来看,三个化合物的半定量分析结果分别比实际浓度高23%、67%和66%,而定量分析结果的偏差仅1%。由此可知,定量分析的结果比半定量分析准确得多。选择定量(完全定量或半定量)方法时应当慎重,在寻求更准确的结果与所需要投入的资源(比如时间和费用)中取得平衡。
耐用性(Robustness)
耐用性是指在测定条件有小的变动时,测定结果不受影响的承受程度。为了使方法可用于日常检验,在分析方法开发阶段,应研究其耐用性,如有苛刻的测试条件,应在方法中注明。对GC/MS和LC/MS而言,典型的变动因素有:待测溶液的稳定性、流动相的组成和pH值、固定相、不同品牌或批号的同类型色谱柱、柱温、流速、进样口和检测器温度等。
溶出物/析出物验证
溶出物/析出物验证是为了考察过滤器和一次性系统在特定工艺中的适用性,以证明其应用不会影响药品的质量和效能,并且不会带来安全风险。该验证过程主要分为以下六个步骤:
(1) 确定直接接触药品的一次性系统;(2) 了解其材料组成、与药品的接触方式、接触条件及生产工艺过程;(3) 对一次性系统进行溶出物试验,根据溶出物预测潜在的析出物,进行风险评估;(4) 用药物产品对一次性系统进行析出物试验,获得析出物数据;(5) 根据析出物数据进行安全性评估;(6) 总结一次性系统对药品的影响,得出适用性结论。
进行溶出试验时,过滤器或一次性系统与试验流体直接接触过程中应尽量保持流体流动,最终获得溶出物抽提液。对得到的溶出物抽提液进行定性和定量分析,从而获得溶出物(即潜在析出物)种类和溶出量。在进行某些分析之前,可能需要对抽提液进行预处理,如前文所述,在预处理时应考察标准品的回收率,以验证预处理方法,保证准确度。
获得溶出物数据后,根据药物生产批量和临床用药的日摄入量,计算出溶出物的日摄入量,再跟日允许暴露量(Permitted Daily Exposure,PDE)等数据进行比较,确认过滤器或一次性系统在工艺条件下产生的溶出物对药物产品是否产生安全风险。
因为析出物通常是溶出物的子集,即溶出物中的化合物种类比相应的析出物多,溶出量比析出量大,因此一般认为溶出物代表了析出物的最差条件。如果溶出物没有超过安全阈值,可以认为该一次性系统适用于该特定工艺条件;如果超过了安全阈值,可以考虑进行析出物试验,结合药品生产批量和药品的每日摄入量,用析出物数据再行评估安全性。
总结
使用一次性系统进行生物制药产品生产,在整个制药行业引起广泛的兴趣和使用热情,有着良好的应用前景。而随着人们对健康和药品质量的日益重视,针对一次性系统的应用可能带来的风险,引起法规部门、标准协会、行业联盟、供应商和终端用户的高度关注,对此展开了深入的讨论和研究,不断出台相关规定、技术文件和解决方案,以推进整个行业的发展。因此,对一次性系统进行溶出物/析出物验证,既符合法规要求,也顺应行业发展趋势。技术上可参考行业推荐的方案和分析方法,对一次性系统的适用性进行全面评估。同时,分析技术的不断发展,痕量物质的分析水平逐步提高,对溶出物/析出物的分析必将越来越精准。随着法规的重视、方法和标准的完善、技术的提高,一次性系统对制药行业必定带来更深远的影响。
【参考文献】
[1] Bio-Process System Alliance, Implementing Single-Use Technology in Biopharmaceutical Manufacturing,Weibing Ding, Jerold Martin, Part One, 2008; Part Two, 2009; Part Three, 2010.
[2] Pall Life Science, Extractables and Leachables from Single-use Systems, USTR 2859, 2012.
[3] Toward Industry Standardization of Extractables Testing for Single-Use Systems: A Collective BPSA Perspective, 2015.
[4] 国家食品药品监督管理总局,药品生产质量管理规范(2010),附录 1,无菌药品。
[5] Bio-Process System Alliance (BPSA) Extractables and Leachables Subcommittee, Recommendation for Extractables and Leachables Testing, Part One, 2007; Part Two, 2008.
[6] 除菌过滤器验证(三):溶出物/析出物,洪海燕,唐燕,2011.
[7] Parenteral Drug Association (PDA), Sterilizing Filtration of Liquids, Technical Report No. 26, 2008.
[8] Parenteral Drug Association (PDA), Application of Single-Use Systems in Pharmaceutical Manufacturing, Technical Report No. 66, 2014.
[9] 化学药品注射剂与塑料包装材料相容性研究技术指导原则,2012.
[10] 中国药典(2010),附录 XIX,A 药品质量标准分析方法验证指导原则。
[11] Standardized Extractables Testing Protocol for Single-Use Systems in Bio-manufacturing, Weibing Ding, 2014.
[12] United States Pharmacopoeia (USP) <233>, Element Impurities-Procedures.
[13] Food and Drug Administration (FDA), Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics, 2014.
彭瑛,唐燕,石茜茹,邢春秀,杨晓慧,李小香
本文由颇尔过滤器(北京)有限公司提供。



![]()
2001-2009Vogel Industry Media版权所有 京ICP备12020067号-15 京公网安备110102001177号

