原研药物表征在仿制药开发中的作用
1984年发布的Hatch-Waxman法案(药品价格竞争与专利期补偿法)中提出的简略新药申请(ANDA),促进了美国仿制药的发展。该法案允许仿制药公司引用原研药物公司在新药申请(NDA)时提交的数据,从而避免了重复的临床试验研究。根据美国FDA要求,在治疗等效性的基础上,仿制药可以替代原研药物。治疗等效性由药学等效性和生物等效性组成,具体解释见下框。
化合物专利一旦过期,一些仿制药公司就可以马上将仿制药投放市场。在高度竞争的仿制药市场,能够首先进入市场是很关键的。仿制药的竞争愈发激烈,与药物价格降低有关。第二个仿制药进入市场时,价格往往会显著降低。因此,仿制药公司必须保证持续新产品上市,以维持收入增长。仿制药公司必须高度擅长于产品和工艺开发,以提供成本可控、具有时效性的产品。
仿制药术语 治疗等效药物:指在相似条件下给药时具有相同的有效性和安全性的药学等效药物。 药学等效药物:含有相同的药物活性成分、剂型、给药途径、规格或浓度的药物制剂。 药学等效不等于治疗等效,因为辅料和/或制备过程中的差异会导致药物性能的差异。药物性能可能在体内或体外环境下存在差异,例如溶出速率和生物利用度。 生物等效药物:在相似试验条件下表现出无显著差异的生物利用度的药学等效药物或药学替代药物。 生物等效性:在一个设计合理的试验研究中,当将两个药学等效药物(或药学替代药物)的药物活性成分(或活性组分)以相同的摩尔剂量给药时,如果两成分在其药物作用部位的吸收速率和吸收程度无显著差异时,则将这两种药物定义为具有生物等效性。 |
一般情况下,仿制药产品开发从对原研药物或参比制剂的表征开始,随后进行药学等效药物设计,开发有效的生产工艺,并开展关键批生物等效性研究。参比制剂表征的内容可多可少,可以是仅仅对药物释放行为(固体口服制剂)或pH和粘度(液体口服制剂)进行检测,也可以是对处方组成进行全面的定量剖析。对参比制剂进行系统、科学的评价,可以得到这些信息。这个过程也被定义为反向工程研究。虽然很多时候仿制药公司都依赖于对原研药物进行表征,但是文献中很少提到这一主题。本章重点从处方和监管的角度总结仿制药开发过程中的原研药物表征。
在药学等效性方面,提升仿制药与参比制剂的处方相似程度,将增加开发出稳定性较好、与参比制剂生物等效的药物的机会。大部分药物产品都是由各种辅料和API组成的多组分剂型,每一种辅料都有各自的作用。虽然在临床上辅料无活性,但在药学方面,辅料具有活性并能影响药物的性能。例如,功能性辅料稳定剂和溶出调节剂,有利于药物的稳定性和生物利用度。某些情况下,即使是稀释剂(例如乳糖或微晶纤维素)等普通辅料也会对工艺性能产生显著影响。
按照美国法律,口服制剂中辅料的定量信息可不披露。在此背景下,对原研药物进行表征是一种加速仿制药开发的科学合理、有成本效益的策略。利用普通文献资源的信息,结合反向工程研究,为开发出在定性和定量方面均与参比制剂相似的处方提供了可能。这为保证仿制药性能提供了更强的信心。
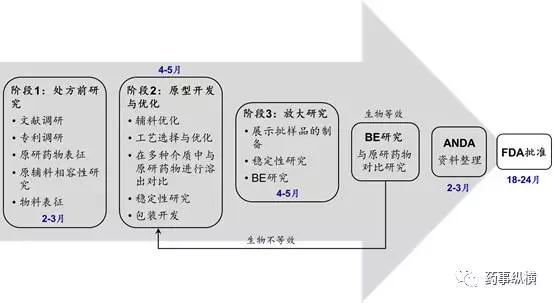
图1 仿制药制剂开发路径图
系统开展的原研药物表征有利于仿制药开发各阶段的决策(图1)。在第一阶段,API固体形式的信息利于识别有价值的技术参数、选择合适的供应商。类似地,为了开发出定性和定量方面均与参比制剂相似的仿制药,需要有一个高度精炼的处方前研究方案。
处方研究的第二阶段将从原研药物表征中得到最关键的信息。关键辅料的定量信息将简化制剂处方优化的工作。传统的处方开发工作包括设计几个处方并考察不同处方的性能和稳定性。解析原研药物的定量信息,可以减少得到最佳处方的实验数量。在质量源于设计(QbD)概念下,原研药物的定量信息有助于识别药物的关键质量属性,从而进行更有效的实验设计。此时的决策对溶出情况等实验结果的依赖性降低,因此决策过程将更客观;溶出情况等实验结果虽然是一个有效的工具,但不能确保一定能开发生物等效的产品。通过原研药物表征,开发出定量方面与参比制剂相似的药物,并保证其与参比制剂溶出行为的相似性,将更容易实现仿制药与参比制剂的生物等效。同样地,对参比制剂中的API进行固态表征,可以降低开发过程中的风险,尤其对于溶出情况决定生物利用度的药物。
通过对参比制剂进行反向工程研究得到的信息,也可以用于支持某些注册申报,例如BE豁免。注射用、眼用、耳用的液体制剂可以BE豁免。关于药物的相似性,美国FDA已经提出了Q1和Q2的概念。Q1是指药物定性相似,而Q2则指药物定量相似。定量相似(Q2)指的是,受试制剂中非活性成分的浓度或用量与参比制剂的差异不超过5%。
辅料的差异一般是允许的,但是监管部门会要求提供额外的理由来证明这些差异不会影响产品的安全性或有效性。按照美国联邦法规,一些特定的剂型具有更严格的要求。在适用于BE豁免的注射用液体制剂仿制药中,除了防腐剂、缓冲剂、抗氧剂之外,其他成分均需符合Q1和Q2。同样地,眼用和耳用液体制剂中,除了防腐剂、缓冲剂、增稠剂或渗透压调节剂,其他辅料均需符合Q1和Q2。局部用药(包括气雾剂和鼻用液体制剂)应符合Q1。这些要求和例外情况的详细信息收录在21 CFR § 314.127(1)(8)和21 CFR § 314.94(a)(9)。
BCS I类的口服速释固体制剂,在完全符合21 CFR § 320.22(b)(3)中规定的各种条件时,也可以BE豁免。当药代动力学终点并不能代表生物等效时,美国FDA也允许对产品进行BE豁免。阿卡波糖片和盐酸万古霉素胶囊是仅仅依据仿制药与参比制剂的Q1和Q2相似性进行BE豁免的两个例子。在这种情况下,对原研药物的全面表征,可用于支持BE豁免。
原研药物表征可以分为四部分:文献分析、处方组成的定量解析、API的固态表征和制备工艺识别。
4.1 文献分析
参比制剂的定性处方组成可以从公开资料中获得。原研公司提交的“批准基础概要”公开部分,是仿制药公司的一个很好的信息来源。这些信息可以在美国FDA网站的“Drug@FDA”板块查阅。一些常规的资料,像产品信息手册和医师案头手册,也包含有用的信息。
4.2 处方组成的定量解析
参比制剂处方组成的定量解析可以进一步细分为两个步骤:1. 关键辅料的识别;2. 对识别出来的辅料进行定量检测。
4.2.1 关键辅料的识别
参比制剂处方组成的定量解析,应该从识别对药物产品稳定性和性能具有显著影响的辅料开始。这些辅料被称为关键辅料。关键辅料包括BCS II类或BCS IV类药物的增溶剂和溶出调节剂,疏水性药物的润湿剂,pH敏感药物的pH调节剂/缓冲剂,易氧化降解药物制剂中的稳定性/抗氧剂。文献报道,山梨醇、甘露醇、甘油和PEGs等一些特定辅料可以通过影响溶解性、溶出速率或渗透性来影响药物的生物利用度。这个过程为后续的原研药物表征提供信息,有助于评估反向工程研究的优势,因为有时传统的处方优化技术可能比反向工程研究更有效。通常,pH调节剂、缓冲剂、稳定剂(例如抗氧剂和螯合剂)、溶出调节剂(例如表面活性剂,控制释放速率的聚合物)最适用于反向工程研究。
4.2.2 对识别出来的辅料进行定量检测
下一步是对固体制剂中的关键辅料进行定量检测,因为制剂中的其他辅料可能会对检测过程有干扰,所以该过程的难度较大。辅料的定量检测涉及两个步骤,即辅料的分离/萃取和辅料的定量检测。以下技术可将辅料从片剂基质中分离出来:溶解度的差异,过滤(使用特定孔径或截留分子量的滤器),高效液相色谱(HPLC),高效薄层色谱(HPTLC),分子排阻色谱。需要结合体系中干扰成分的数量和及其理化性质来选择合适的分离技术。
辅料分离以后,可以采用重量法或各种检测工具进行检测,检测工具包括紫外-可见光、折光率、蒸发光散射检测器(ELSD,HPLC)、光谱技术(例如红外(衰减、透光率、反射系数)或近红外光谱)。重量法最适用于制剂中用量较大的主要辅料的定量检测。稳定剂、表面活性剂和pH调节剂等用量较小的辅料最适用于复杂的分离和定量技术,例如HPLC和HPTLC。聚合物等高分子量辅料可以采用分子排阻色潽法进行有效检测。
其他有用的技术包括气相色谱、液相-质谱、胶束电动毛细管色谱、比色法、电位测定法、变角衰减全反射傅立叶变换红外光谱、核磁共振、毛细管电泳。检测过程需要注意消除假阳性和假阴性结果。针对制剂中其他辅料可能存在的干扰,检测方法必须进行验证。最近,检测仪器领域有了一些新的进展,例如,马尔文的SyNIRgi近红外化学成像系统宣称可以检测制剂中每一种辅料的用量。
从药学角度看,固态性质可以分为分子水平、颗粒水平、粉末整体水平的性质(图2)。分子水平的性质包括晶型、水合物、溶剂化物、共晶和无定型态。分子间排列和自由能的差异使得这些形式具有不同的溶解性、可制造性、生物利用度和稳定性。仿制药开发过程中,当对参比制剂中API固态形式进行表征时,这些因素是关键的参数。在其他关于ANDA的文献中,已对多晶型进行了详细的阐述。根据目前法规要求,法规条文并不要求仿制药公司证明“仿制药中的API与参比制剂具有相同的物理性质,药物固态形式未改变”。因此,药物的固态多晶型并不是证明ANDA中药物相似性的一个相关项目。
原研药物一般选择最稳定晶型进行制剂产品开发,以避免工艺过程和储存过程中的固态形式转变。安全起见,仿制药公司也使用与参比制剂相同的晶型,以确保稳定性和溶出特征与参比制剂相似。有时候,因为晶型专利的限制,这种策略是行不通的,晶型专利的期限长于化合物专利。在此情形下,可根据505[j][2][A][vii],选择其他晶型的原料药开发仿制药。各种技术均可用于表征药物的固态形式,例如XRD、FTIR、NIR、拉曼光谱、DSC、热重分析和热台显微镜。API颗粒粒径分布(PSD)可能影响溶出速率和生物利用度,尤其对于生物利用度受溶出影响较大的药物。可以检测原研药物和仿制药制剂产品中的API颗粒粒径分布。但是,在各种工艺条件下,API的初始粒径可能会发生变化,例如,(i)湿法制粒过程中API的溶解,随后在干燥过程中沉淀;(ii)混合/过筛过程中颗粒粒径减小;(iii)压片过程中发生破碎。当药物制剂中的API颗粒粒径分布发生变化时,需改变API初始粒径分布的控制标准。

图2 不同水平的固态性质及其重要性
对制剂产品中API的粒径检测的挑战是,在其他辅料存在的情况下检测API颗粒粒径。基于遮光和激光散射的常规颗粒粒径检测技术不适用,因为这些技术不能区分API和辅料。唯一可行的技术是显微镜检查。
基于颗粒形状和双折射模式等特征,显微镜检查可区分API和辅料。在偏振光下,晶型药物表现出双折射特性,而大部分辅料是非晶态的、不表现出双折射特性。基于熔点的差异,热台显微镜可识别API。因此,从分子和颗粒水平对API进行识别并表征,可以加快制剂开发过程中的决策。虽然没有相关的报道,采用光谱成像技术检测颗粒粒径分布在理论上是可能的。
根据API稳定性、API在片中所占的重量比和物理性质(例如,流动性和可压缩性),大部分固体口服制剂是采用湿法制粒、干法制粒或直接压片工艺制备的。除了加工性能之外,制备技术也会影响药物制剂的稳定性和性能。
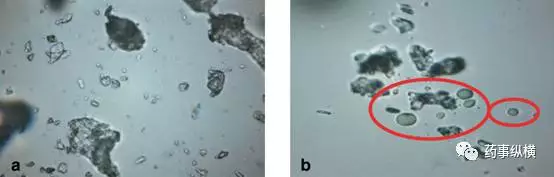
根据API理化性质,可以预测参比制剂的制备工艺,例如,湿法制粒工艺不会用于水敏感性API。类似地,对于低剂量API,采用直接压片工艺很难实现合适的混合均匀度。目测观察药片的断裂情况,可以得到一些制粒工艺方面的信息。湿法或干法制粒工艺的药片断裂面比直接压片更粗糙。或者,可将药片放入培养皿中的水里,使用低功率光学显微镜观察崩解特性(图3)。直接压片制备的药片,崩解成为单个的粒子(particle),而湿法或干法制剂工艺制备的药片崩解为粒子团块(granule)。该信息可与定性的处方组成信息结合起来分析,确定剂型中各辅料的作用。有些辅料在剂型中可能发挥多种作用,例如羟丙甲纤维素、淀粉和乳糖。因此,仅仅通过定性的处方组成,很难确定各辅料的功能。
先进的成像技术,例如SEM、光谱成像(拉曼、NIR、兆赫兹)、XRF,可以检测药片和丸的包衣厚度和/或组成。对于含有功能包衣的产品,这是很有价值的信息。
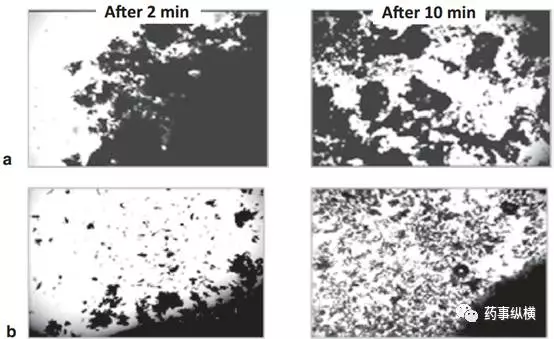
图3 药片在水中2 min、10 min时崩解方式的显微照片(使用光学显微镜观察,400×)。a:制粒工艺;b:直接压片
参比制剂表征过程中会遇到各种挑战。通过理解物料特性、选择合适的样品制备和分析方法,可以降低参比制剂表征的复杂性。但是,由于药物制剂本身的复杂性,这些挑战不可能完全避免,例如:
1. 药物含量低(例如API占总片重的比例低于5%)
2. 药物颗粒的粒径非常小(例如,药物颗粒粒径接近亚微米)
3. 辅料晶型的复杂性(例如,制剂中含有一些晶型/非晶型辅料)
4. API的固态形式转变(例如,API是亚稳态晶型,工艺过程/溶剂/温度可能导致其晶型发生转变)
5. API和辅料的双折射性质、熔点、溶解度等理化性质及其相似
6. 复方制剂
图4所示是片剂原研药物表征的决策树。辅料的功能性和原料药的理化性质决定参比制剂表征的难度。
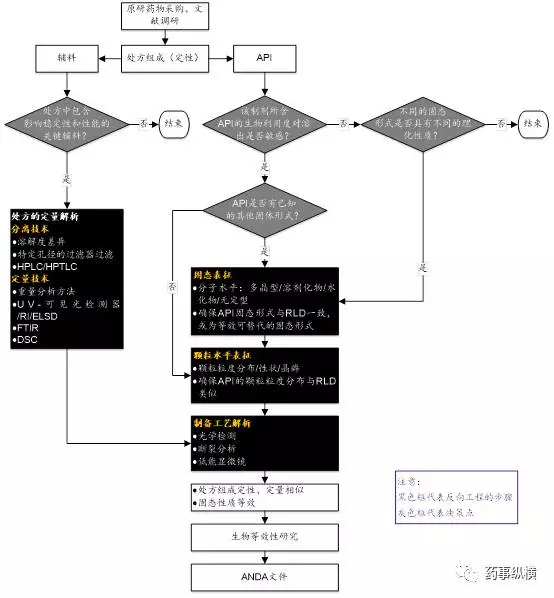
图4 片剂反向工程研究的决策树
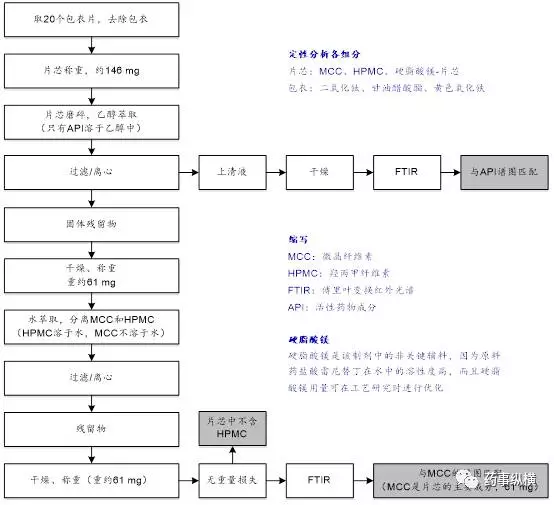


液体制剂是一种或多种化学物质溶于或分散于合适的溶剂系统中形成的均匀分散体系。根据给药途径,可以将液体制剂分为以下几类:口服,注射(静脉、肌肉、皮下注射),耳用,眼用,局部使用。口服给药的液体制剂包括溶液剂、糖浆剂、乳剂、混悬剂。为了避免过度复杂,在此我们简要介绍口服溶液剂。液体制剂经常包含结构和浓度差异很大的多种辅料,使得体系更加复杂。因此,对一种处方进行解析可能和设计一个新的产品具有相当的挑战性。
如上所述,对参比制剂的解析,始于鉴别能够改善API溶解度和稳定性的辅料。溶剂中溶解的溶质总量一般远低于其饱和溶解度。因为API以溶解状态存在于体系,API的固态形式不影响液体制剂的稳定性和性能。在液体制剂中,API的固态形式、颗粒特征等物料属性会影响工艺参数,例如溶出速率。
第二步是对参比制剂中使用的辅料进行定量测定。与传统的方法(检测药物和辅料在不同比例下的相容性)不同,反向工程研究显著减少试验数量,有助于优化最终产品。通过调节pH实现原位成盐、使用共溶剂、络合作用,可以用于对API的增溶作用。此外,液体制剂中可能包含调节pH的缓冲剂、保持化学和物理稳定性的稳定剂、抑制微生物生长的防腐剂,以及改善病人顺应性的矫味剂和着色剂。如果药物具有苦味,液体制剂中一般会添加掩味剂。与片剂不同,溶液的制备工艺并不太复杂。工业规模时,溶液剂的制备是通过机械搅拌的方式、在大的混合容器中将药物和非活性成分简单混合。
依据 21 CFP § 320.22(b)(3)(i),口服溶液剂(酏剂、糖浆剂、酊剂)、注射剂(IV-静脉、IM-肌肉、SC-皮下)和其他局部用溶液剂的体内生物利用度合格,可以BE豁免。其前提是溶液剂中不含有显著影响药物吸收的辅料,且药物制剂中的药物释放是没问题的。只要生物利用度未改变,缓冲剂、防腐剂和抗氧剂允许存在差异。但是,溶液的pH需要和参比制剂保持一致。因此,对于溶液剂而言,对其处方组成进行定量检测确实是一种性价比高、节省时间的方法。
成本和产品上市速度是仿制药公司的成功关键。实现与参比制剂生物等效,是仿制药开发的关键环节,一定要降低生物不等效的机率。全面系统的原研药物表征是仿制药开发的有效工具,可以增加生物等效的概率。合理可靠的原研药物表征策略(对原研药物进行反向工程研究),包括处方组成的定量检测、API的固态表征和制备工艺识别,可以缩短产品开发周期、降低成本。
文章来源:药事纵横
热点文章
-

2025版药典有哪些内容变动
2024-09-23
-

三种常见制粒技术对中药制剂内在质量的影响及生产过程控制要点
2024-09-27
-

科普 | 新药研发全流程梳理(图文版)
2024-12-03
-

中国药典2025版带来的冲击和影响
2024-10-04
-

洁净工作服清洗、灭菌及使用效期验证
2024-10-14
-

药品检查过程中关于偏差管理的分析与研究
2024-10-15
-

小组件 大作为:细胞和基因疗法 (CGT)工艺的闯关秘籍
2024-12-03
-

固体制剂高活性车间设计策略
口服固体制剂作为临床应用非常广泛的剂型之一,其传统生产模式存在产尘量大、生产暴露环节众多以及工序复杂等特点。因此,在生产 OEB4-5 级标准的口服固体制剂时,面临的挑战是多方面的。本文从车间建设的角度出发,探讨了针对高毒性或高活性等固体制剂生产所需采取的技术手段与措施。
作者:
-
降本增效的能源管理实施建议
-
直播预约 | 小核酸大未来:小核酸商业化生产与厂房设计建设
-
技驭未来,揭秘民营企业科技创新的基因密码
-
小柴胡颗粒连续逆流动态提取工艺研究
-
注射用甲苯磺酸奥马环素无菌检查抗菌活性的去除及验证





评论
加载更多