
压缩气体的微生物监测
对于洁净室洁净空气设备监测粒子和微生物污染,制药行业遵循以下法规:
欧盟GMP Annex 1,2008版;
FDA无菌生产指导方针;
USP/EP 污染限度的要求;
中国GMP 附件1,2010修订版;
当行业指导方针中没有明确规定特定测试方法,很重要的是去看ISO标准中的方法。
各类指导方法并不制定具体的空气或表面等的微生物和粒子洁净度的确定方法.需要参考其他国际性的文件比如EN/ISO标准等。(以上在EU GMP Annex 1 2008中提及)。
当确认压缩空气的微生物监测的测试方法时,可以参考ISO 8573-7压缩空气—第7部分:活性微生物污染的测试方法。这个标准在这篇文件内也将再次提到。
从主管机构比如FDA 21CFR 211制剂的现行良好生产规范中也可以看到直接的要求:在药品生产的生产,加工,包装及转运阶段中,应当采用可充分控制压差,微生物,微尘,湿度和温度的仪器。——21CFR 211.46(b)
在FDA的行业指导方针-无菌工艺生产的无菌药品—现行的良好生产规范中要求所使用的压缩空气应当等同于或者由于所进入环境的空气质量。压缩空气应当具有适合的纯净度(比如无油)和它过滤后的微生物和粒子质量应当等同于或者优于所进入环境的空气质量。
因此,在欧盟GMP的A级环境,中国GMP的A级环境或者FDA的关键环境的气体应当不超出所在环境等级的粒子和微生物的限度。
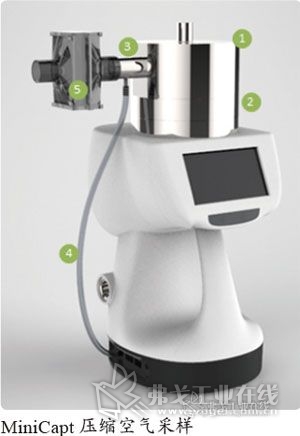
采样方法
通常要求日常测试和监控压缩空气,并需要符合现行的制药生产的法规。
具体如何做,可以查询ISO 8573-7的指导方法。ISO文件包括了区分活性,菌落群,微生物与其他可能在压缩空气中存在的固态粒子的测试方法。这个标准提供了一个采样,培养和确定微生物粒子数量的手段。测试方法适合于确认符合ISO 8573-1的洁净等级,同时也可用ISO8573-4鉴别固态粒子是否是活性的,成菌落团存在的。
这个方法在压缩空气采样时采用一个促生长的培养基,平皿或者类似材料。要求在使用狭缝式撞击仪器采样同时采用减压气体。气体的流速需要记录,并且需要在不超过4h的时间段内测量;然而,大多数培养基的干燥率都要求暴露时间控制在90min以内。
测试方法要求:
所有设备在使用前灭菌处理;
连接采样进气口与压缩空气管路,包括任何与设备连接电子控制元件;
在测试时设备先空吹,可以让压缩空气将设备内任何残留的消毒剂吹干净,这个测试先不放培养基平皿;
在可旋转的升降平台上放入90mm培养皿;
取下培养皿的盖子,盖上MiniCapt的采样头,然后将培养皿的盖子放入无菌袋中;
培养皿可调整高度至适合于MiniCapt采样器的位置;
开启设备,并设置能够在90min采样左右采样到1m3的采样参数;
在采样结束时,拿出无菌袋子里面的培养基盖子,打开MiniCapt采样器上面的采样头,并将培养基盖子盖上暴露的平皿;
取出培养基平皿,记录采样参数,将平皿在适合的温度和必要的时间内培养,最后记录菌落数。
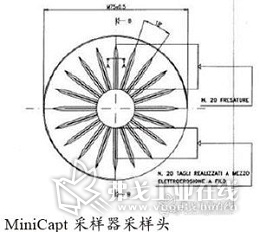
测试采用MiniCapt压缩空气配件配合MiniCapt环境采样器操作。这个方法采用了20条辐射状狭缝撞击减压后的空气至培养基上。培养基高度可通过采样器的升降平台进行调整,使培养基与采样头的空间达到最理想的采样效率,符合ISO 14698的浮游菌采样物理采样效率和生物采样效率的相关要求。一旦培养基已经培养于规定的时间段内,培养出的菌落数和菌落形态即可读出,并可对比欧盟GMP,中国GMP和FDA的洁净室法规的限度要求。测试应当以一定频率重复进行,这样能够最好的反映制剂生产时的风险。这反过来也反映了压缩空气所进入的洁净室的等级级别。
A级和FDA关键区应当每生产班次至少监测1次;
B级和FDA支持区应当每天1次;
C级每周1次;
D级每月1次。


![]()
2001-2009Vogel Industry Media版权所有 京ICP备12020067号-15 京公网安备110102001177号


加载更多