
o-RABS/c-RABS与无菌药品的生产控制

无菌药品种类繁多,治疗用途广泛——其中注射剂,吸入剂,眼用制剂,创伤类外用药占据无菌制剂的前四位。本文简述o-RABS和c-RABS原理,比较设计和功能的异同、进展及其在无菌药品生产中的应用。
文/陈洪,汪磊
目前批准上市的绝大部分小水针、冻干、大输液等无菌制剂的生产环境要求B/ISO7级洁净环境下,直接与药品接触的高风险灌装、压塞等核心工序则要求在A/ISO5级单向层流保护下进行生产。随着微球/脂质体,胶束,纳米粒等特殊注射剂(DDS)的开发上市,RABS作为设计观念领先、方便实用的无菌生产专用设备,无论是从生产条件控制、洁净级别,还是人/料分离操作,都有着单元操作性强、体积小、总体生产成本低、适于中小批量生产等特点,备受厂家的青睐,现就RABS的类型、功能和无菌生产控制等关键问题进行论述。
定义和概念
RABS(Restricted Access Barrier System)概念的提出始于上世纪90年代Upjohn的Stewart Davenport,国内RABS和隔离器这两个概念有时通用,需注意区别(下文详述)。虽然设备体系用于无菌生产或检验已经有20多年的历史,但目前仍然缺乏一个权威统一的定义,文献报道中的含义也不尽相同。国际商业化的隔离器,其不同品牌在性能上也有差别。总体而言,o-RABS/c-RABS的字面含义为限制进入的生产隔离系统,其中o是open,c是closed,分两种类型,前者指开放式设备体系,后者指封闭式体系。RABS作为先进的生产系统体现了人物分离和防止污染的理念,综合International Society for Pharmaceutical Engineering(ISPE)对RABS的宽泛表述,将其要点概括为4个方面:
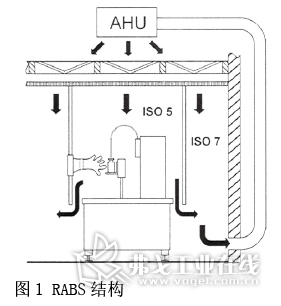
● 关键工艺在A/ISO5级区,RABS原理图(见图1);
● 生产过程最大限度避免了人的干预;操作者通过手套箱或半身无菌服操作;
● 进入舱室的物料和物件/配件等经过消毒和灭菌除热源,避免受到人及环境的二次污染;
● 舱室内的生产环境-生产线可清洁灭菌和接受杀孢子剂消毒,灌装/转运/传递的工器具等可进行灭菌除热源(与传统开放式B级洁净室相比较,RABS优势明显)。
ISPE为此专门成立了RABS专业委员会,成员来自FDA,国际药企和设备厂家,定期讨论RABS发展态势并对行业规范进行更新。
RABS材质一般由316L、聚碳酸酯或玻璃、硬质塑料或软质塑料(如聚氯乙烯)建成。结构包括空气处理系统HEPA、传递接口及传递门、灭菌气体发生器、配套设备与辅助设施等四部分,其中物料及配件在单体设备之间或多台RABS间传递可通过特殊设计的RTP(快速传递门)进行递送和控制,RTP通过互锁环或法兰叠合,以密封圈封闭,防止微生物进入舱内。
o-RABS采用独立HEPA及HVAC系统(被动式RABS的进风系统非专用),舱内空气排入B级洁净室内,采风部分或全部来自室内,所以舱内温湿度及单向层流受到室内空气影响,舱内空气不可以循环无助于降低HVAC能耗,且不可以生产毒性大/高活产品,对操作者防护低。
c-RABS介于o-RABS向隔离器过渡的设计,有独立的HEPA和HVAC,舱内空气可处理后循环使用,且不排放入B级背景区。舱内空气维持正压,可进行毒性级别较高的药品生产(排放到大气的粉尘和空气需经过处理达到排放标准),舱内温湿度采用专用HVAC可精确控制;缺点为对环境操作者保护不足,根据EHS及设备泄露率设计考察,极高活品种不适于生产。目前新一代c-RABS吸取了Isolator的优点,可在线清洁和灭菌,但对设备的设计和制造,材质和加工性能和精度等提出了较高的要求,整体造价也大大超过o-RABS。企业可以根据产品特点和风险评估结果,灵活选择合适用途的设备。进入中国的外资品牌有IMA、Bosch、Telstar等7家,国内也有多家推出不同设计类型的RABS(见图1)。
隔离器Isolator与RABS有本质的区别,从设计理念讲,隔离器是更成熟可控的无菌生产体系。隔离器与环境全隔离,设备所在区域可为C级,独立的HEPA和HVAC,系统防泄漏率达到ISO10648-2,对环境和操作者安全友好,可进行高活性和OEB5类产品生产。
无菌药品的生产控制
对于无菌注射剂,特别是F0<8的产品,只能采用非最终灭菌方式,给产品的生产和无菌保障(SAL)带来很大的挑战,RABS成为特殊无菌制剂的首选设备(目前的c-RABS的设计和应用在向isolator发展)。理论上说,现有无菌生产车间及设备设施都存在的设计瑕疵或缺陷,这是产品生产面临高风险和高污染的主要原因,无菌生产采用RABS需要注意以下几点:
(1)操作者是无菌药品生产的主体,尽量减少人与物料及设备配件表面的直接或间接接触,这是无菌药品生产设计的关键。除非在检修或停产阶段,任何侵入舱体的操作都应避免;在生产中发生故障的短暂处理,需要进行风险和措施的二次污染评估。
(2)保持舱内的正压;减少和控制不合理的舱内操作,不影响A级下气流的风型。
(3)对于特殊注射剂,需要合理优化工艺流程,尽量减少灌装前的时间,RABS的上游单元提供了料液浓配、稀释、过滤、除热源、料液传递、灌装、压塞,甚至微球的干燥,乳剂的冻干等都可以实现在多台RABS里集成,最大限度的减少人为因素或二次污染。
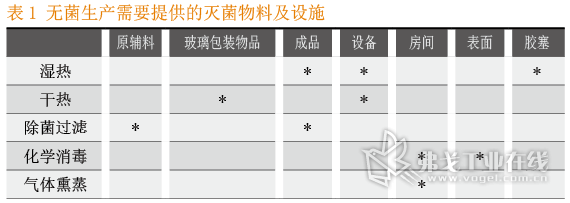
(4)进入舱内物件,生产线材质可耐受化学杀孢子剂以去除芽孢,或采用流通蒸汽及干热灭菌等适宜方式去除热源,见表1。
(5)对于封闭式隔离器不直接与外界环境相连,使用无菌接口或快速转移通道RTP进行物质传递,开放式隔离器允许材料通过舱门进人,内部保持正压,阻止微生物的进入。
(6)API的除热源,料液灌装前如何保障不被二次污染,这是工艺设计的核心。尤其对于先进的DDS产品,大多为溶解度低,对湿热不稳定,API和高分子载体如卵磷脂等不能耐受湿热/干热/紫外灭菌,对除热源和生产过程中不被热源污染提出挑战;对于批量小的微球或脂质体品种,常采用一次性配置容器,进行物料/料液的配置,配置成适宜浓度,并过滤除菌;一次性容器体积达到50 L,内设搅拌装置,可以实现温度的调控和取样检测,并留有物料递送接口;配有专门的过滤除菌和除热源装置,但目前国际公认,对于除热源,非破坏性或非侵入性的除热源方法如超滤和阴阳离子树脂吸附法等尚不能完全适应商业化,而且除热源的效率不高;对于注射剂除热源,国际上已明确摒除活性炭吸附法,所以物料的转运和生产过程的热源控制成为无菌工艺验证的关键环节,尤其起始物料的热源必须满足1 EU/ml以下。0.22 μm滤器的完整性验证包括起泡点、扩散流或压力保持实验,但无法去除热源和病毒/支原体,这是目前过滤器设计缺陷。同时滤芯材质对API的吸附和相容性,细菌截留挑战,流量流速,压力,滤膜上下压差,过滤时间,初始物料微生物负载,以及过滤循环次数等都需要在工艺中验证考察或设计挑战性试验,以确定边界参数,制定合理的工艺参数范围。
小结
综上,随着无菌制剂新剂型新功能辅料的应用,与批量和剂型特点相适应的生产设备RABS越来越受到关注。合理筛选和使用RABS的功能,发挥其在产品无菌控制,降低二次污染和风险的作用,需要从产品自身和RABS提供的优势性能两方面入手,确保从源头上进行无菌工艺的合理设计,为患者提供安全可靠疗效确切的优质产品。
【参考文献】
[1] 2015版中国药典:无菌检查用隔离系统验证指导原则.
[2] Pharmaceutical Engineering,2005,Vol5,No6.


![]()
2001-2009Vogel Industry Media版权所有 京ICP备12020067号-15 京公网安备110102001177号


加载更多