
制药企业GMP有效执行的探讨
当前,随着我国GMP 法规与国际标准接轨,药品生产质量管理水平得到了很大的提升——但需要清醒地认识到,GMP 在推行过程中仍然存在质量管理理念还有待加强、没有形成质量文化、技术方面还存在墨守成规,以及内部管理缺少有效的检查和监督等诸多问题。本文从写你所做、做你所写、记你所做和改你所错4 个方面论述GMP 执行过程中可能遇到的主要问题,总结提出相应的改善措施,以促进GMP 法规在制药企业更加有效地执行。
自1999 年开始从国家层面在药品生产领域强制推行药品生产质量管理规范(GMP)至今已有22 年,2011 年3 月1 日起执行新版GMP(2010 年修订版)已有10 年。我国GMP 法规已与国际标准欧盟GMP 接轨,使得我国药品生产质量管理水平得到了很大的提升,但需要清醒地认识到,GMP 在推行过程中仍然存在质量管理理念还有待加强、没有形成质量文化、技术方面还存在墨守成规,以及内部管理缺少有效的检查和监督等诸多问题。本文通过分析探讨GMP 执行过程中可能遇到的主要问题,总结提出相应的改善措施,以促进GMP 法规在制药企业更加有效地执行。
什么是GMP
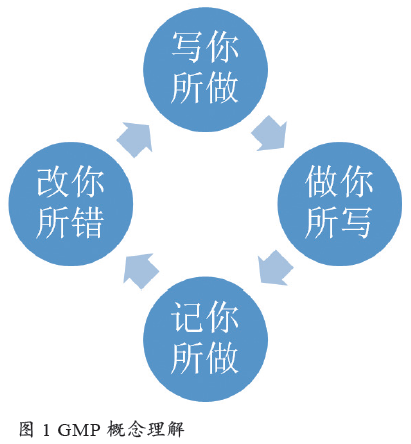
GMP 的中文名称是药品生产质量管理规范,是质量管理体系的一部分,可以将GMP 的概念理解为:写你所做,做你所写,记你所做,改你所错( 如图1所示)。“写你所做”,统筹布局建立执行标准;“做你所写”,照章办事强调不折不扣执行标准;“记你所做”,数据完整以便追溯检查;“改你所错”,总结提升不断修正改进。GMP 的执行也是从建立标准、执行标准、形成记录到改进标准之后再到建立标准的循环过程,在实践过程当中使自身质量管理水平得到有效提升。
执行GMP 可能存在的主要问题及分析
写你所做
(1)文件体系抄袭套用,未结合企业实际情况及最新法规要求。
(2)当岗位实际操作与文件体系规定不一致时,未及时修订。
(3)管理人员起草编写文件时,岗位操作人员未参与文件编写及修订,以致文件规定与实际情况脱节。
(4)文件审核把关不严,部分文件本身不具备可操作性。
GMP 检查缺陷举例:公司未按照计算机化系统验证与确认及验证附录要求,开展相关培训、文件制定及实施工作。
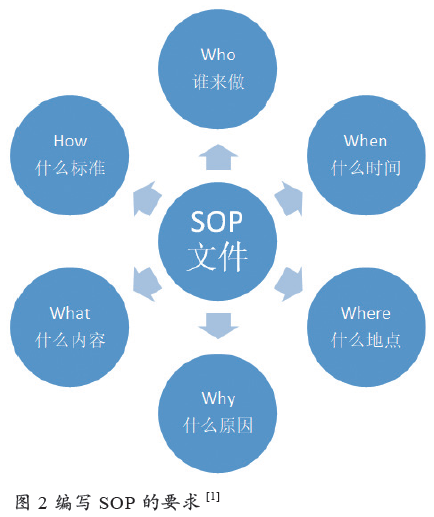
GMP(2010 年修订版)第八章对文件管理方面作了详细规定。公司GMP 文件系统编写体现了管理层合法依规意识和技术团队的技术管理水平,好的SOP 文件必须满足5W1H 要求(如图2 所示)。文件是确保信息有效传递和管理的工具,从高层重视提供资源,营造合规质量文化,再到全员统一思想,结合实际讨论达成共识、关注法规和标准及时更新文件系统,进而调动一线员工积极参与,不断检查纠正,在制度层面对体系进行风险管理,最终做好GMP有效执行方面的顶层设计。
做你所写
(1)文件执行前未进行实质培训,或未评价培训效果,未确认实操能力。
(2)岗位操作过程中不关注文件,由管理人员现场指挥替代。
(3)岗位所需文件不便取阅或未发放。
(4)岗位操作人员未真正理解文件并未按文件要求逐条执行。
GMP 检查缺陷举例:文件的批准、复制、分发、销毁未按规程进行管理,无复制、分发、替换或销毁记录,《原辅料供货商代号表》无文件编号、未经质量部门批准,生产部门提供了两个版本的《地巴唑片工艺规程》,其中旧版(STP-GY-016-R00)已失效,现行版(STP-GY-016-R01)未受控。
GMP 第184 条规定:所有药品的生产和包装均应当按照批准的工艺规程和操作规程进行操作并有相关记录[2]。遵照规程操作是每个员工的基本素养和要求。管理人员要深入生产一线了解文件系统的执行情况,定期开展文件规定与执行情况一致性的检查,关注一线员工对文件内涵理解情况,这也是对质量管理体系运行有效性的检验和监督。
记你所做
(1)人员未行成及时准确完整填写记录的习惯, 数据完整性存在问题。
(2)记录设计存在缺陷,不方便填写,未合并重复事项。
(3)提前写记录及事后补记录。
(4)记录填写的方式不统一,归档之前未严格审核,归档之后不便查阅。
GMP 检查缺陷举例:检验记录涉嫌造假,仪器使用日志不真实。在该企业液相工作站打印出的精制冠心片药粉液相色谱图,批号为20130301、20140501、20140801、20140802、20150901样品液相色谱图高度一致,涉嫌一图多用的数据完整性问题。
记录是GMP 执行留下的证据。GMP 要求记录要做到及时、准确、真实、完整和按规定修改,这需要通过检查和培训对企业员工进行引导。记录的设计在符合法规要求的同时要简洁优化,避免重复填写。为避免填写过多文字性内容,应多采用固化内容之后进行勾选。另外定期归档记录时要审核检查,及时纠正日常工作当中存在的问题。
改你所错
(1)文件执行与记录填写的错误重复出现,未落实整改。
(2)长期存在的问题,未找到根本原因并制定解决措施。
(3)检查监督未全面覆盖并有效落实。
(4)管理人员未真正认识到质量管理过程中存在的失误。
(5)对发生的变更未进行变更控制,使风险增大。
GMP 检查缺陷举例:胞磷胆碱钠注射液质量标准(含量测定)项下要求进样量为10μL,实际操作进样量为20μL,企业未启动偏差调查,而是将原始检验记录中高效液相色谱图的进样量手工修改为10μL,且企业审核未发现该问题。
有的企业称其企业管理体系运行良好,没有偏差也没有变更,甚至有的管理者对指出其管理职责内存在的问题比较抵触,不能以良好的心态接受并进行更正。这极不可取,对于反复出现的较高风险偏差,一定要组织技术人员专题讨论攻关,采取适宜的纠正和预防措施进行管控。
改善方法探讨
每个公司都有不同的组织架构与管理流程,但GMP 管理实质是相通的,人员是软硬件结合实施的主体,是工作质量的决定者。GMP 执行首先要考虑加强人员和内部体系管理,改善方法建议如下:
决策层树立正确导向,营造企业自律氛围公司决策层是企业发展的掌舵者,也是各项措施落实情况的监督检查者。GMP(2010 年修订版)第4条规定:企业应当坚持诚实守信,禁止任何虚假、欺骗行为。这个导向一定要正确,制药企业必须严守诚实守信的底线,任何时候不能突破。企业决策层要知法、懂法、用法,意识和理念要随着GMP 发展而更新,在企业营造严格自律遵守法律法规的氛围,重认证轻管理、重硬件轻软件、重效益轻人才“三重三轻”的思想观念一定要转变[3],在内部管理上层层落实责任制,让各种有限的资源得到合理的分配应用。各级管理人员不能出现只报喜不报忧,管理人员的职责应像漏斗,而不是过滤器,实事求是地传递信息才能让大的风险得到及时管控。关注质量体系有效性,重视技术团队建设
1998 版GMP 更关注符合性,2010 版GMP 更关注有效性,我们要认识到符合不一定有效,在执行GMP 的过程中,很多药厂往往是法规要求有的文件都有,但是未在实际当中运用并形成实效[4]。文件是企业的行为准则,首先要组建技术过硬的团队,结合公司实际情况,把文件体系建立好,需重点考虑内容合规性、格式术语统一性、可操作性、与实际一致性、引用唯一性、流程简捷,以及全员参与等编写原则,文件体系的编写情况体现技术团队的技术实力与管理水平,也是体系有效性的基本保障,与药品的质量直接关联。
在技术团队管理方面,应建立制度使组织利益与个人利益相融合,如果组织目标的实现与个人的利益关系不大, 就很难把组织目标与个人目标结合在一起[5]。核心技术人员管理是企业管理的关键,吸引并尊重高层管理人员并给予较高的福利待遇,可以在企业实行职业技术等级考评,建立良好的班组文化精益管理;不同部门管理人员应进行阶段性换岗学习、实行质量考核制度等,强化责任意识,完善监督检查;不同员工应有差异化的管理,与工资福利绩效考核挂勾。管理人员既要有良好的工作规划,也要深入一线进行检查监督,检查计划的落实情况和文件要求的执行情况,发现实际存在的问题并及时组织解决。
深刻理解产品风险管控,深入调查研究科学决策
制药企业对产品生产、储存、运输、使用全过程进行风险管控,先进行正确的风险识别,从而决定正确的风险管控措施。必须及时全面查找出产品高风险事项,对那些发生频次高、较难发现、后果严重的事项实施重点风险控制精准管理,分析和研究相关数据,制定相应的CAPA 措施。
科学运用质量工具,为GMP有效执行提供保障,不能将风险管理只是简单地运用风险管理工具撰写出风险评估报告,而是要基于团队经验和对产品质量、工艺的深刻理解和执行情况,撰写风险评估报告。作决策前一定要开展风险评估,使决策更科学合理和具有可操作性。
重视偏差管理不断改进,鼓励因地制宜不断创新
任何偏离程序和标准的情况均应纳入偏差管理,偏差管理是GMP 执行过程中重要的环节,其作为一种发现问题、分析问题、解决问题并持续改进质量管理体系的有效手段,对提升质量管理理念、提高质量改进的执行力具有重要意义[6]。如果想真正提升企业的执行水平,就要认真关注每一个异常,组成多专业团队人员一起去认真分析,不断寻找症结所在,从根本上解决问题,这是提升GMP 管理及执行水平最有效的途径。
根据企业的实际情况,不断创新管理方式,才能适应法规环境的不断变化。例如, 创新检查方式,在企业内部实行飞行检查、突击检查等,在检查中融入新的检查手段,采用横向拉网、纵向突破的检查方式[7],使GMP 进入常态化管理;创新培训方式,充分利用网络资源进行移动课堂培训和考核,达到系统性拓宽知识面的目的,使优秀的技术人员进行深入学习从而获得提升。
GMP 的有效执行没有句号,只有逗号。制药企业要在满足法规要求的同时,结合实际情况努力提升执行效率和药品质量。制药人要始终不忘药品生产质量管理治病救人的初心,牢记全心全意为人民服务的使命,不断探索共同推动医药行业更加合规有序、平稳健康地发展。
【参考文献】
[1] 汪达. 浅谈新版GMP 文件系统编写[J].机电信息,2012(05):24-28.
[2] 国家药品监督管理局. 药品生产质量管理规范.2010 年修订版.
[3] 谢敬东,李野,杨亚明,秦晓瑞. 论提高我国医药生产企业GMP 管理执行力的对策[J]. 中国医药导报,2008(16):105+116.
[4] 汪达. 浅析质量意识缺失存在的风险.[J].机电信息,2013(29):25-28.
[5] 张来俊. 执行新版药品GMP 不能“三重三轻”[J]. 首都医药,2012,19(17):18-19.
[6] 汪达, 张宝月, 龙华燕. 药品生产偏差管理探讨[J]. 化工与医药工程,2019,40(04):61-64.
[7] 刘忠兴,侯宝亮. 确保GMP 制度有效执行的思考[J]. 首都医药,2007(12):11.


![]()
2001-2009Vogel Industry Media版权所有 京ICP备12020067号-15 京公网安备110102001177号


加载更多